



Introduction
Deep vein thrombosis (DVT) is a blood clot that forms within a deep vein, especially in leg veins such as the femoral vein. The prevalence of DVT is slightly higher in men than in women [1] and increases with age [2]. Approximately 20%–50% of patients after a first DVT event suffer from post-thrombotic syndrome (PTS), which involves intractable edema, pain, a sensation of heaviness, pigmentation, and even venous ulcers in severe cases [1, 3]. In addition, DVT can be potentially life-threatening when a blood clot breaks off and develops into pulmonary embolism (PE), leading to sudden cardiovascular collapse and death [4, 5]. DVT and PE form a single disease process named venous thromboembolism (VTE), which is the third most common vascular disease in the United States [6, 7]. The main consequences of DVT are death, recurrence, PTS, and major bleeding caused by anticoagulation [5, 8]. Consequently, the quality of life of patients with DVT is severely affected, especially when PTS is present [5, 9]. Approximately 900,000 new cases of DVT are estimated each year in the United States alone, with a mortality rate close to 300,000 cases per year [10, 11]. Therefore, it is important and necessary to study the molecular mechanism of DVT and to explore alternative therapies.
Neovascularization is a major event for the resolution of thrombus [12, 13]. A growing body of data has suggested that bone marrow-derived circulating endothelial progenitor cells (EPCs) are recruited into the thrombus and accelerate thrombus resolution and also have an important role in physiological and pathological neovascularization in adults [13–16]. In addition, EPCs have the ability to proliferate, migrate, and form new blood vessels through differentiation into endothelial cells, and have recently become a promising therapeutic approach for DVT-related thrombus resolution in patients who have achieved limited success with current treatment options [13, 16, 17]. Although EPCs show good therapeutic effects, their clinical application still faces many challenges. The number and function of circulating EPCs can be affected by adverse conditions in the microenvironment, including advanced age, diabetes, cardiovascular risk factors, ischemic disease, and graft vasculopathy [18–21]. Therefore, the development of methods to improve the recruitment of EPCs to the thrombus and enhance angiogenesis is needed.
Long non-coding RNAs (lncRNAs), a class of non-coding RNAs longer than 200 nucleotides, regulate molecules through multiple mechanisms including epigenetic modification, as well as transcription and translation regulation [22, 23]. Studies have also shown that lncRNAs can serve as competitors or reservoirs of miRNAs to indirectly regulate gene expression [24], and lncRNAs are emerging as crucial regulators of angiogenesis, development, differentiation, metabolism, and autophagy [21, 25–28]. Autophagy, one of the crucial degradation pathways in eukaryotes, is important for maintaining cellular homeostasis and is associated with vascular diseases [29]. Furthermore, multiple lncRNAs are dysregulated in various diseases including vascular disease and may control disease progression, providing promising new targets for the treatment of vascular diseases [30, 31]. However, the roles of lncRNAs in DVT recanalization and resolution are still largely unknown.
We previously performed a comprehensive expression profiling analysis of lncRNAs in DVT patients, and found that lncRNA GUSBP5-AS (enst00000511042) was significantly upregulated in EPCs from DVT patients compared with healthy controls [32]. GUSBP5-AS is located at chromosome 4q31.21 on the somatic map with a length of 342 bp. We suspected that GUSBP5-AS may modulate angiogenesis in EPCs and participate in the dissolution of thrombi. This article focuses on the role of GUSBP5-AS in EPCs and in DVT resolution, and explores the underlying molecular mechanism. These findings may contribute to the development of a novel effective treatment for DVT.
In this study, we demonstrated that GUSBP5-AS was upregulated in EPCs in DVT patients. Overexpression of GUSBP5-AS in EPCs significantly reduced thrombus size and weight and facilitated the homing ability of EPCs to DVT sites to promote DVT recanalization and resolution. Furthermore, we found that GUSBP5-AS promotes angiogenesis and cell proliferation, migration, and invasion, and inhibits apoptosis in EPCs by modulating fibroblast growth factor 2 (FGF2) and matrix metalloproteinase-2/9 (MMP2/9) expression through the miR-223-3p/FOXO1 (Forkhead Box Protein O1)/Akt pathway. These findings may provide new insights into the molecular mechanisms of lncRNAs in regulating angiogenesis and suggest a potential novel therapeutic approach for DVT.
Results
GUSBP5-AS facilitates EPC homing to thrombosis sites and DVT recanalization and resolution
EPCs can be recruited to sites of thrombosis and accelerate vasculogenesis and thrombosis resolution [33]. We next investigated whether GUSBP5-AS regulates the homing ability of EPCs to thrombotic sites and participates in DVT recanalization and resolution. We constructed a model of jugular vein thrombosis in nude mice and injected GFP-GUSBP5-AS-EPCs or GFP-NC-EPCs, as a control, to thrombi. The thrombosis size in the GFP-GUSBP5-AS-EPC group was smaller than that in the LV5-NC group (Figure 1A). In the GUSBP5-AS overexpression group, the thrombus weight was significantly reduced on day 7 after transplantation compared with the LV5-NC group (Figure 1B). In addition, the number of EPCs homing to thrombosis sites in the GUSBP5-AS-overexpressing EPC group was higher than that of the LV5-NC group (Figure 1C–1D). Immunofluorescence analysis demonstrated that there were higher expression levels of CD34, a marker of angiogenesis in the thrombus, and MMP2, an essential regulator of early thrombolysis, and more nucleated cells including EPCs in the thrombi of the GUSBP5-AS overexpression group compared with the LV5-NC group (Figure 1E). Together, these results indicated that GUSBP5-AS accelerates DVT revascularization and resolution and may represent a novel potential target for DVT treatment.
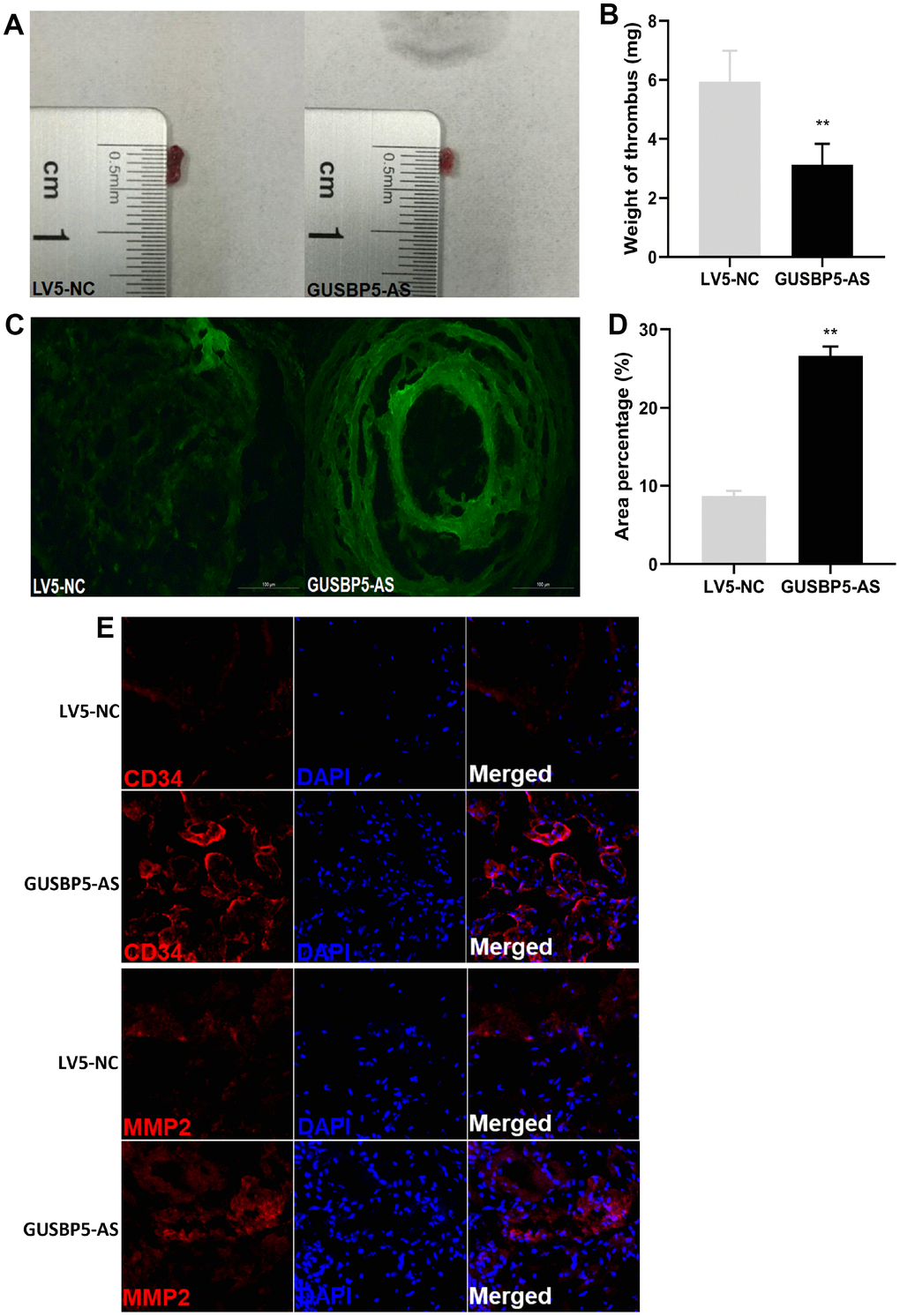
Figure 1. Effects of lncRNA GUSBP5-AS on the treatment of DVT. (A) Representative images of the thrombosis in GFP-GUSBP5-AS-EPC group and LV5-NC group were observed. (B) Weight of the venous thrombi at day 7 after the transplantation. Data are expressed as mean ± SEM (n = 8 mice per group), **P < .01 compared to the corresponding negative controls. (C) Representative images of recruitment of GFP-positive EPCs in DVT (×200). (D) Percentage of green fluorescent area in DVT. **P < .01. (E) The expression levels of CD34 and MMP2 in thrombosis were detected by immunofluorescence.
GUSBP5-AS enhances the angiogenesis of EPCs in vivo and in vitro, migration, invasion, F-actin filaments and proliferation, and inhibits cell apoptosis
To examine the effects of GUSBP5-AS on EPCs, we performed gene knockdown and overexpression by lentiviral infection and observed EPC angiogenesis in vivo and in vitro, migration and invasion. The results of angiogenesis in vivo assay showed that implants with GUSBP5-AS-overexpressing EPCs were redder in appearance than the LV5-NC group, while the implants with GUSBP5-AS knockdown EPCs were less red compared with those of the LV3-NC groups (Figure 2A). No differences were observed between the LV5-NC group and LV3-NC group, and the Matrigel alone group (without cells) was not red in appearance. H&E staining revealed more luminal structures in implants with GUSBP5-AS-overexpressing EPCs than in those with LV5-NC EPC group and fewer luminal structures in implants with GUSBP5-AS-knockdown EPCs than in in those LV3-NC EPCs; no luminal structures or cells in Matrigel implants without cells were observed (Figure 2B). To confirm our in vivo results with EPC function and angiogenesis, we next investigated the role of GUSBP5-AS in angiogenesis in EPCs in vitro. The tube formation of GUSBP5-AS-overexpressing EPCs was significantly increased compared with the LV5-NC group, while angiogenesis in GUSBP5-AS-knockdown EPCs was attenuated compared to the LV3-NC group (Figure 2C). These results demonstrated that GUSBP5-AS is functionally involved in EPC-associated angiogenesis. To examine the role of GUSBP5-AS in cell migration, we performed wound healing assays and observed that migration of EPCs with GUSBP5-AS-knockdown was significantly lower than that of the LV3-NC group while the migration ability of the GUSBP5-AS overexpression group was significantly greater than that of the LV5-NC group (Figure 2D). Transwell invasion assay showed similar results; inhibition of GUSBP5-AS significantly reduced cell invasion, while overexpression of GUSBP5-AS enhanced cell invasion (Figure 2E). We further studied whether GUSBP5-AS acts on cell cytoskeleton that is closely related to changes in cell migration capacity. As shown in Figure 2F, downregulation of GUSBP5-AS impaired F-actin filaments while GUSBP5-AS overexpression prevented disruption of the actin cytoskeletal structure. These results revealed that GUSBP5-AS plays a central role in the migration and invasion ability of EPCs in vitro, likely by regulating the expression of F-actin, at least in part.
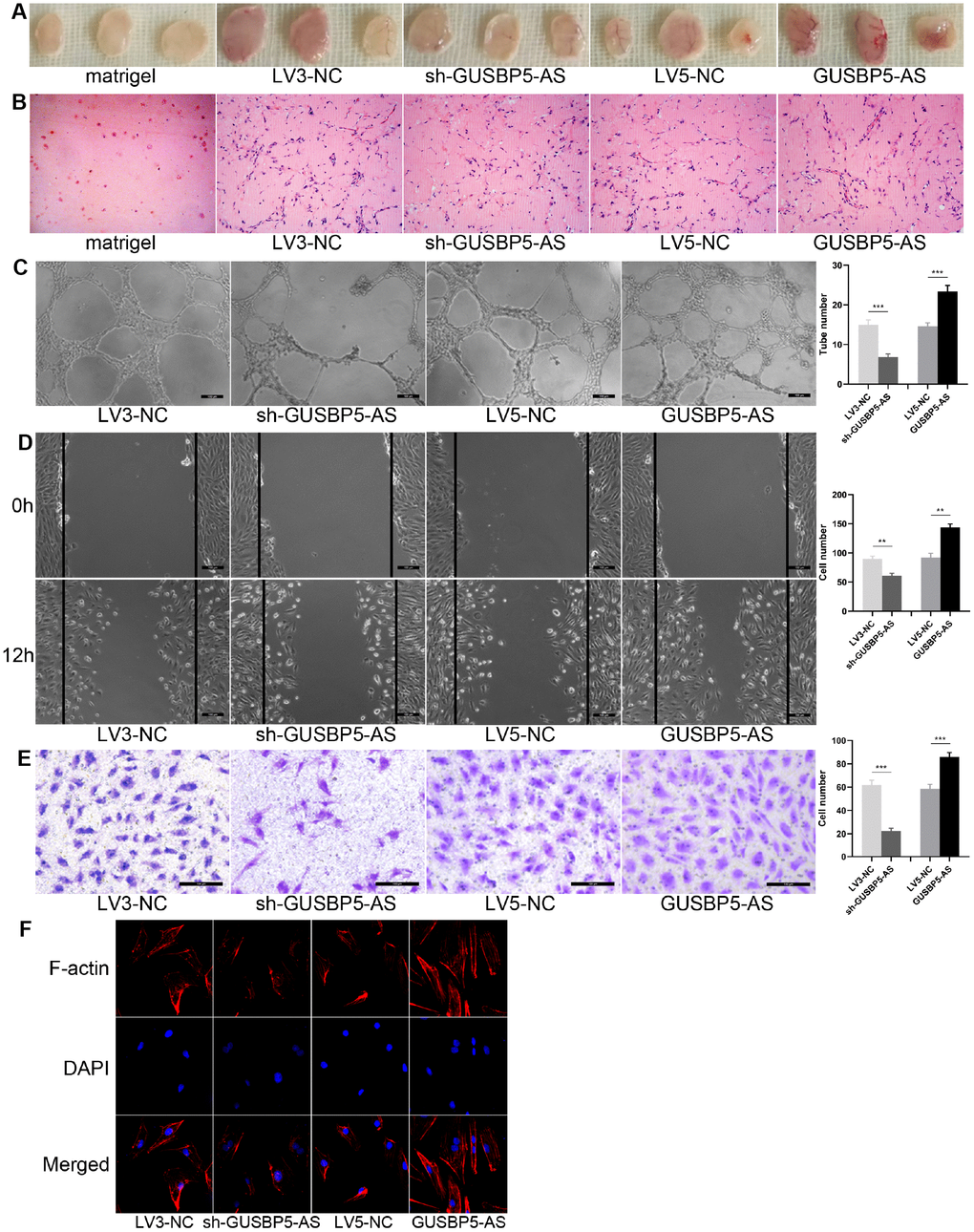
Figure 2. Effects of lncRNA GUSBP5-AS on angiogenesis of EPCs in vivo and in vitro, EPC migration and invasion. (A) In vivo angiogenesis was evaluated at Day 7 after subcutaneous injection of Matrigel-mixed EPCs into nude mice. (B) HE staining: lncRNA GUSBP5-AS knockdown and overexpression, respectively, decreased and increased tube formation by EPCs in vivo (original magnification, ×200). (C) In vitro tube formation assay. Scale bar=100μm. (original magnification, ×100). LncRNA GUSBP5-AS knockdown and overexpression, respectively, decreased and increased tube number. ***P< .001. (D) Wound healing assay showing the effects of lncRNA GUSBP5-AS on EPC migration. Scale bar=100μm. (original magnification, ×100). LncRNA GUSBP5-AS knockdown and overexpression, respectively, decreased and increased cell migration in EPCs. **P< .01. (E) Effects of lncRNA GUSBP5-AS on cell invasion ability were analysed. Scale bar=100μm. (original magnification, ×200). Data are represented as mean ± SEM. ***P< .001. (F) Effect of lncRNA GUSBP5-AS on actin cytoskeleton structure in cultured EPCs. Cells were fixed, permeabilized, stained with rhodamine–phalloidin and DAPI, and visualized by confocal microscopy; down-regulation of GUSBP5-AS impaired F-actin while GUSBP5-AS overexpression prevented disruption of actin cytoskeletal structure.
GUSBP5-AS have a central role in EPC growth
We next performed CCK8 assays to evaluate the role of GUSBP5-AS in regulating EPC proliferation. Cell proliferation was increased in the GUSBP5-AS overexpression group compared with controls, while the proliferative ability of EPCs in the GUSBP5-AS-knockdown group was decreased compared with the LV3-NC group (Figure 3A). Changes in cell proliferation are usually associated with changes in cell cycle progression. To study the mechanism by which GUSBP5-AS regulates EPC proliferation, we performed cell cycle analysis on EPCs with GUSBP5-AS overexpression and knockdown and their corresponding control groups. Flow cytometry analysis revealed that overexpression of GUSBP5-AS increased the proportion of EPCs arrested in G1/S phase but decreased the portion in G2/M phase compared with the LV5-NC group. GUSBP5-AS inhibition significantly reduced the proportion of cells in S phase compared with the LV3-NC group (Figure 3B). These results suggested that GUSBP5-AS exerts an important role in cell cycle regulation, especially in S phase. We next investigated whether GUSBP5-AS regulates apoptosis in EPCs. Flow cytometric analysis of treated EPCs showed that GUSBP5-AS overexpression significantly decreased the percentage of total apoptotic cells, while inhibition of GUSBP5-AS significantly promoted apoptosis, especially for early apoptotic cells (Figure 3C). Taken together, our data demonstrated that GUSBP5-AS has a critical function in the regulation of in vivo and in vitro angiogenesis of EPCs, migration, invasion, and proliferation, and inhibits cell apoptosis.
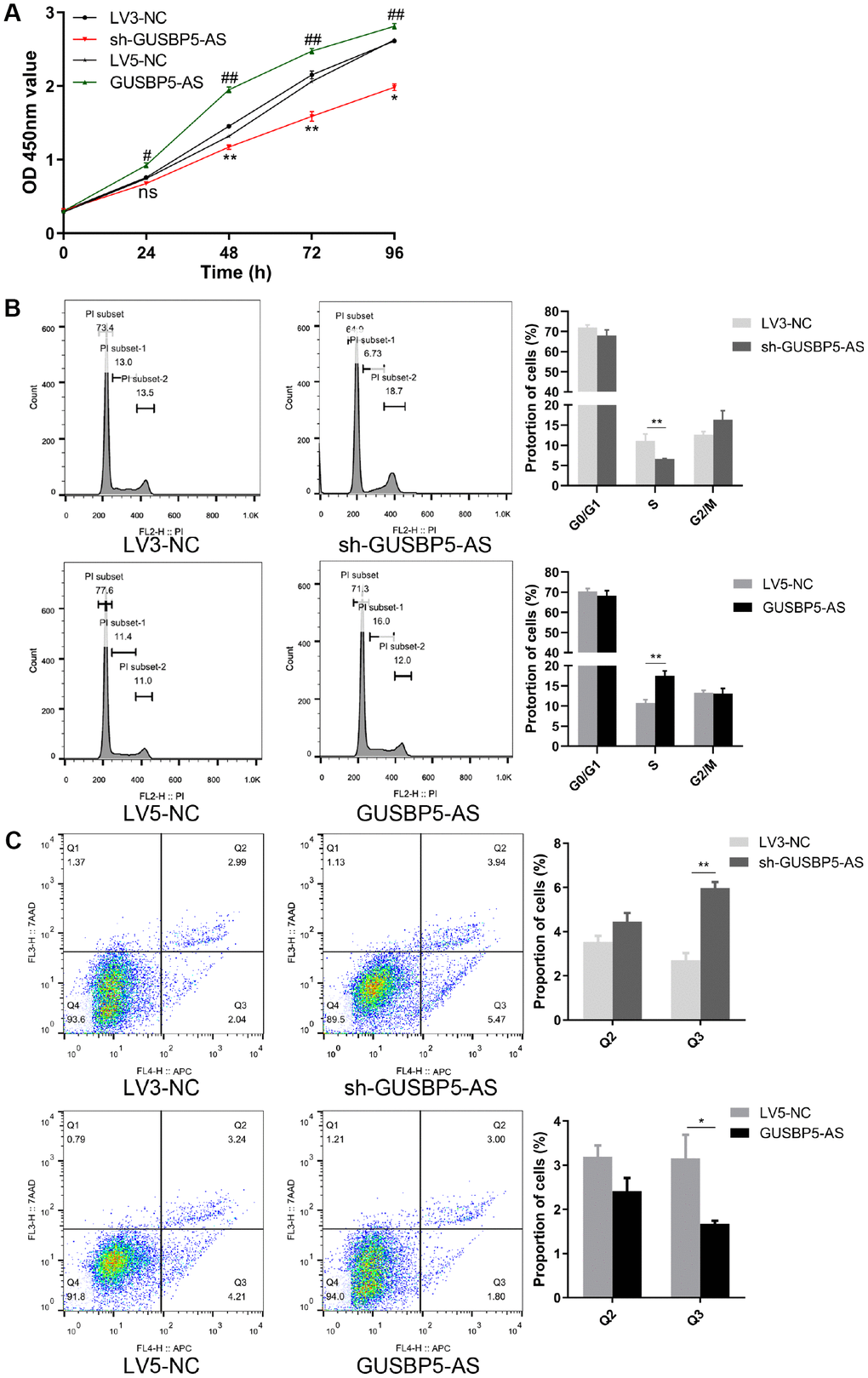
Figure 3. GUSBP5-AS have a central role in proliferation. (A) Cell proliferation ability of GUSBP5-AS in lncRNA GUSBP5-AS knockdown and overexpression group and their corresponding NC groups was detected by CCK8 assay. *P< .05, **P< .01 compared to the LV3-NC group; #P < .05, ##P < .01 compared to the LV5-NC group. (B) The effect of GUSBP5-AS on EPC cycle was investigated by FCM cell cycle distribution test. (C) The effect of lncRNA GUSBP5-AS on cell apoptosis was measured with Annexin V-APC/7-AAD Apoptosis Detection Kit by FCM. Cell apoptosis was analysed. Data from 3 experiments are summarized. *P< .05, **P< .01 compared to the corresponding negative control group. Data are represented as mean ± SEM.
GUSBP5-AS is closely related to the regulation of EPC function by RNA sequencing
For validation purposes, we next performed RNA sequencing (RNAseq) to evaluate gene expression profiles in GUSBP5-AS-overexpressing EPCs by microarray analysis. mRNA expression levels were screened in GUSBP5-AS-overexpressing EPCs and negative controls using hierarchical clustering analysis (Figure 4A). The results showed that 2,430 (52%) genes were upregulated and 2,220 (48%) genes were downregulated in the GUSBP5-AS overexpression group compared with the control group (Figure 4B). To identify relevant biological functions, Gene Ontology (GO) enrichment analysis of Biological Processes (GOBP) was performed. Genes modulated by GUSBP5-AS showed a significant involvement mainly in categories related to metabolic processes, stress, cell cycle, and gene expression (Figure 4C). Following verification of the regulatory role of GUSBP5-ASin vivo and in vitro, we next investigated the underlying mechanism of GUSBP5-AS in the modulation of angiogenesis and migration in EPCs. As shown in Figure 4D, lncRNA-miRNA-mRNA network showed that 12 predicted miRNAs including miR-223-3p may be targets of GUSBP5-AS. Based on the results of mRNAseq and the regulatory network of lncRNA-miRNA-mRNA, we used qRT-PCR to detect the expression levels of all the predicted target miRNAs (Data were not shown), and found that miR-223-3p expression was significantly decreased by GUSBP5-AS whereas downregulation of GUSBP5-AS significantly increased the expression levels of miR-223-3p. Moreover, the main genes involved in angiogenesis and migration that are significantly altered by GUSBP5-AS are shown in Table 1 and Supplementary Figure 1. The results of RNAseq and qRT-PCR suggested that the main factors, FOXO1, MMP2, MMP9 and FGF2, were upregulated and inhibited, respectively, by GUSBP5-AS overexpression and knockdown.
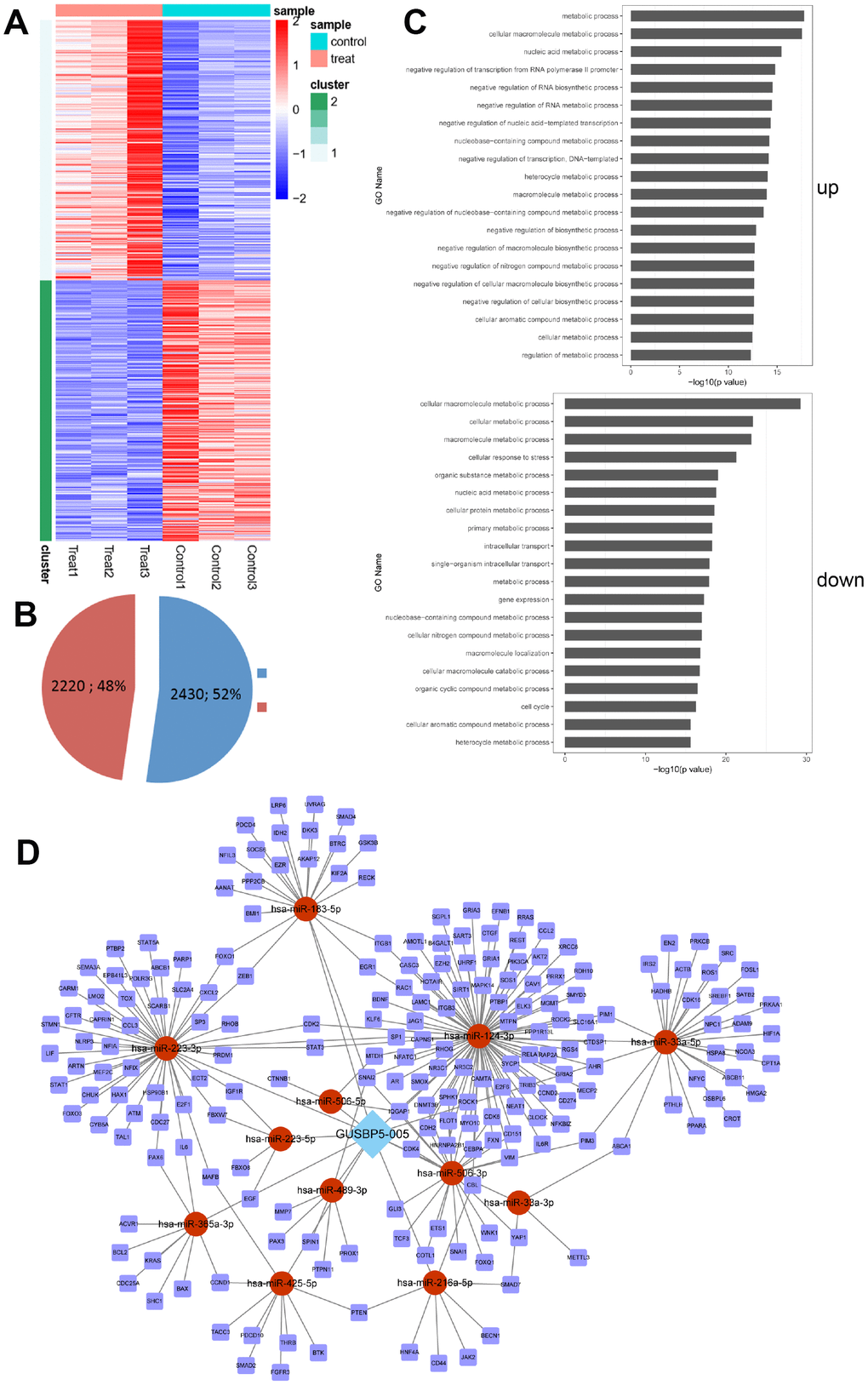
Figure 4. The up- and down- regulated mRNAs were filtrated using microarray analysis. (A) Heatmap analysis of dysregulated mRNAs in negative controls and GUSBP5-AS-overexpressing EPCs. Control group indicates LV5-NC group and treat for GUSBP5-AS-overexpressing EPC group. (B) Proportion of up- and down- regulated genes were filtered using microarray analysis in the GUSBP5-AS overexpression group compared with the LV5-NC group. Blue indicates up-regulated genes, and red for down-regulated genes. (C) Several biological process pathways were enriched. Up indicates the enrichment analysis of up-regulated genes, and down for down-regulated genes. (D) The regulatory network of lncRNA-miRNA-mRNA.
Table 1. RNAseq analysis results of the main factors involved in angiogenesis and migration regulated by GUSBP5-AS overexpression.
| Gene_id | Log2FoldChange | Stat | p value | p adj | Significant | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMP9 | 1.680414704 | 4.19413 | 2.74E-05 | 0.000111 | Ups | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FGF2 | 1.381545685 | 3.44619 | 0.000569 | 0.001783 | Ups | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| MMP2 | 1.22681 | 2.06987 | 0.0008 | 0.005605 | Ups | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| FOXO1 | 1.232851707 | 5.50857 | 3.62E-08 | 2.35E-07 | Ups | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| RNAseq analysis was performed after EPCs infection with LV5-NC and GUSBP5-AS lentivirus, and three groups of LV5-NC and three groups of GUSBP5-AS were detected; Gene id means differentially expressed genes; p value was calculated by t test, and p adj was obtained by p value normalized by Benjamini-Hochberg; significant means the up- or down-regulation of the gene expressed in the GUSBP5-AS overexpression group compared with the LV5-NC. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
GUSBP5-AS directly binds miR-223-3p to regulate migration and angiogenesis in EPCs
To further verify the binding relationship between miR-223-3p and GUSBP5-AS, bioinformatics prediction tools was used. These data also revealed that miR-223-3p has complementary binding sites that target the 3’ untranslated region (3’ UTR) of GUSBP5-AS (Figure 5A). Dual luciferase reporter assay confirmed that miR-223-3p binding to GUSBP5-AS displayed reduced luciferase activity, indicating molecular interactions (Figure 5B–5D). These data indicated that GUSBP5-AS directly sponges miR-223-3p. As shown in Figure 5E, GUSBP5-AS suppressed miR-223-3p expression, and the expression of miR-223-3p increased following overexpression of miR-223-3p. These results indicated that miR-223-3p is a downstream molecular target of GUSBP5-AS. To further explore the function of GUSBP5-AS and miR-223-3p in EPCs, migration, invasion, and tube formation experiments were performed. Overexpression of GUSBP5-AS and miR-223-3p in EPCs significantly increased and decreased tube formation, migration and invasion, respectively, compared with the NC group (Figure 5F–5H). We did not observe differences between the GUSBP5-AS and miR-223-3p mimics cotransfection group and the NC group.
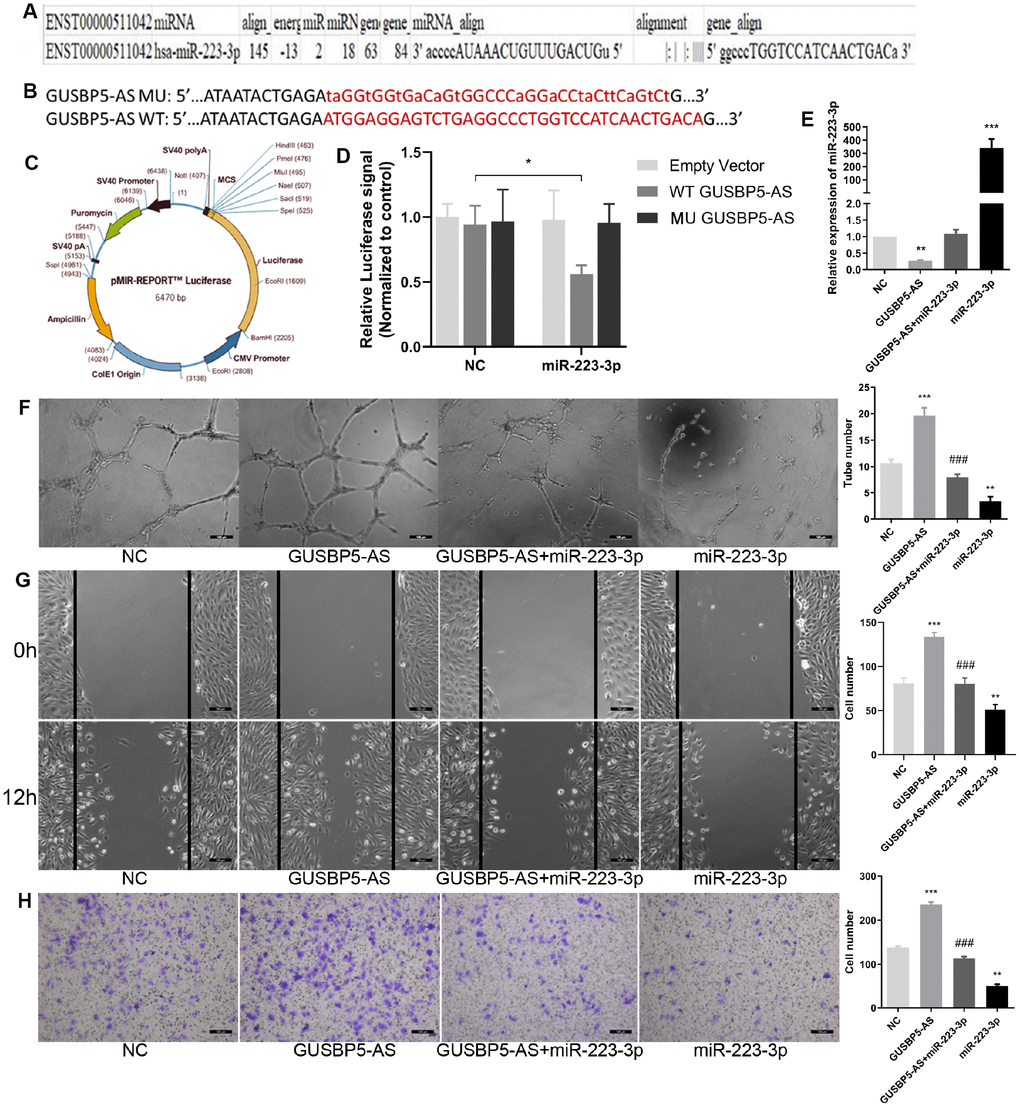
Figure 5. LncRNA GUSBP5-AS directly bound with miR-223-3p. (A) Bioinformatics analysis revealed GUSBP5-AS contains binding sequences complementary to the binding sites of miR-223-3p. (B) The luciferase reporter constructs containing WT-GUSBP5-AS or MU-GUSBP5-AS sequence. (C) Schematic graph of the constructed reporter plasmid containing putative. (D) Dual-luciferase reporter assay verified the targeting relationship between miR-223-3p and GUSBP5-AS. (E) The expression of miR-223-3p was measured by qRT-PCR after cells treated with GUSBP5-AS, GUSBP5-AS+miR-223-3p or miR-223-3p. (F) Effects of cotransfection of GUSBP5-AS and miR-223-3p on in vitro tube formation were analysed. Scale bar=100μm. (original magnification, ×100). (G) Effects of cotransfection of GUSBP5-AS and miR-223-3p on cell migration were detected by wound healing assay. Scale bar=100μm. (original magnification, ×100). (H) Effects of cotransfection of GUSBP5-AS and miR-223-3p on cell invasion were analysed by transwell cell invasion assay. Scale bar=100μm. (original magnification, ×100). **P < .01, ***P < .001 compared to the NC group, ###P < .001 compared to the GUABP5-AS treated group. Data are represented as mean ± SEM.
FOXO1 is a direct target of miR-223-3p in EPCs
To investigate the mechanism of miR-223-3p in angiogenesis, the downstream target genes of miR-223-3p were predicted using the Starbase, TargetScan, miRanda, and miRDB databases. According to RNAseq analysis, the regulatory network map and predicted targets of miR-223-3p (Table 1 and Figure 4D), FOXO1, playing a key role in cell function, may be a potential target gene of miR-223-3p, and the binding sequences between miR-223-3p and FOXO1 mRNA are shown in Figure 6A. FOXO1 is a member of the FOX transcription factor family, which is involved in numerous crucial cellular processes including metabolism, proliferation, and migration, as well as cell cycle, apoptosis, and angiogenesis progression [34, 35]. Previous studies showed that FOXO1 expression can be regulated by miRNAs. To examine the association between miR-223-3p and FOXO1, luciferase reporter plasmids of miR-223-3p and FOXO1 were used (Figure 6B). Reduced luciferase reporter activity was observed after cotransfection of the luciferase reporter plasmid containing WT-FOXO1 and miR-223-3p mimics in HEK-293T cells (Figure 6C). Moreover, miR-223-3p mimics downregulated FOXO1 mRNA expression while miR-223-3p increased its expression in EPCs (Figure 6D). Furthermore, to further verify the correctness of our results, we tested the role of FOXO1 in the regulation of EPC function. As showed in Figure 6E–6G, knockdown of FOXO1 significantly inhibited the abilities of EPC angiogenesis, migration and invasion, whereas FOXO1 overexpression obviously enhanced EPC function. These results indicated that miR-223-3p targets and negatively regulates FOXO1 in EPCs.
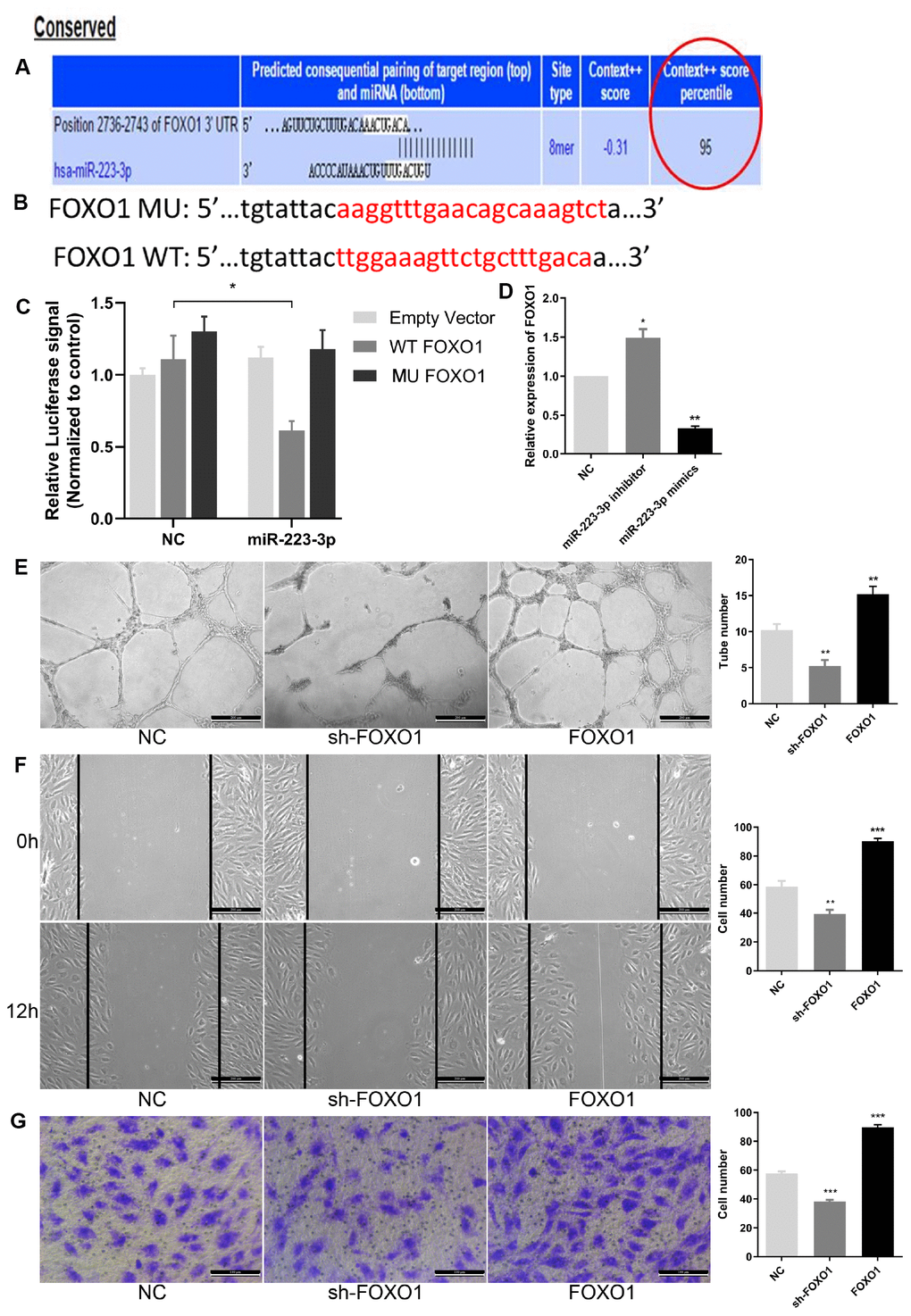
Figure 6. FOXO1 acted as the direct target of miR-223-3p. (A) Bioinformatics programs assay revealed complementary binding within miR-223-3p and FOXO1 3’UTR. (B) The luciferase reporter constructs containing WT-FOXO1 or MU-FOXO1 sequence. (C) Dual-luciferase reporter assay verified the targeting relationship between miR-223-3p and FOXO1. (D) Relative quantification of mRNA levels of FOXO1. Three independent experiments were performed. (E) Tube formation assay showing the angiogenesis ability of EPCs by FOXO1 knockdown and overexpression. Scale bar=200μm. (original magnification, ×100). FOXO1 knockdown and overexpression, respectively, repressed and enhanced capability of angiogenesis in EPCs. (F) Effects of FOXO1 on cell migration were analysed by wound healing assay. Scale bar=200μm. (original magnification, ×100). FOXO1 knockdown and overexpression, respectively, reduced and increased number of EPCs migrated. (G) Transwell invasion assay showed that FOXO1 knockdown and overexpression, respectively, inhibited and promoted the ability of cell invasion in EPCs. Scale bar=100μm. (original magnification, ×200). **P < .01, ***P < .001 compared to NC group. Data are represented as mean ± SEM.
The relationship between GUSBP5-AS, miR-223-3p and FOXO1 in EPCs from DVT patients
As shown in Figures 5E and 6D, GUSBP5-AS overexpression significantly downregulated miR-223-3p expression and miR-223-3p inhibitor observably increased the expression levels of FOXO1 in EPCs by qRT-PCR assays. To test the relationship between GUSBP5-AS, miR-223-3p and FOXO1 in EPC samples from DVT patients, the Spearman rank correlation analysis were performed by qRT-PCR. The data showed that miR-223-3p expression was negatively correlated with GUSBP5-AS and FOXO1 expression, while GUSBP5-AS expression was positively correlated with FOXO1 expression (Figure 7A–7C). Additionally, we verified the expression levels of GUSBP-AS and FOXO1 in EPCs infected with miR-223-3p and GUSBP-AS, respectively. The results showed that miR-223-3p mimics and inhibitor significantly suppressed and enhanced GUSBP5-AS expression respectively, and the expression of FOXO1 was notably decreased and increased, respectively, by GUSBP5-AS knockdown and overexpression, compared to the corresponding controls (Figure 7D–7E).
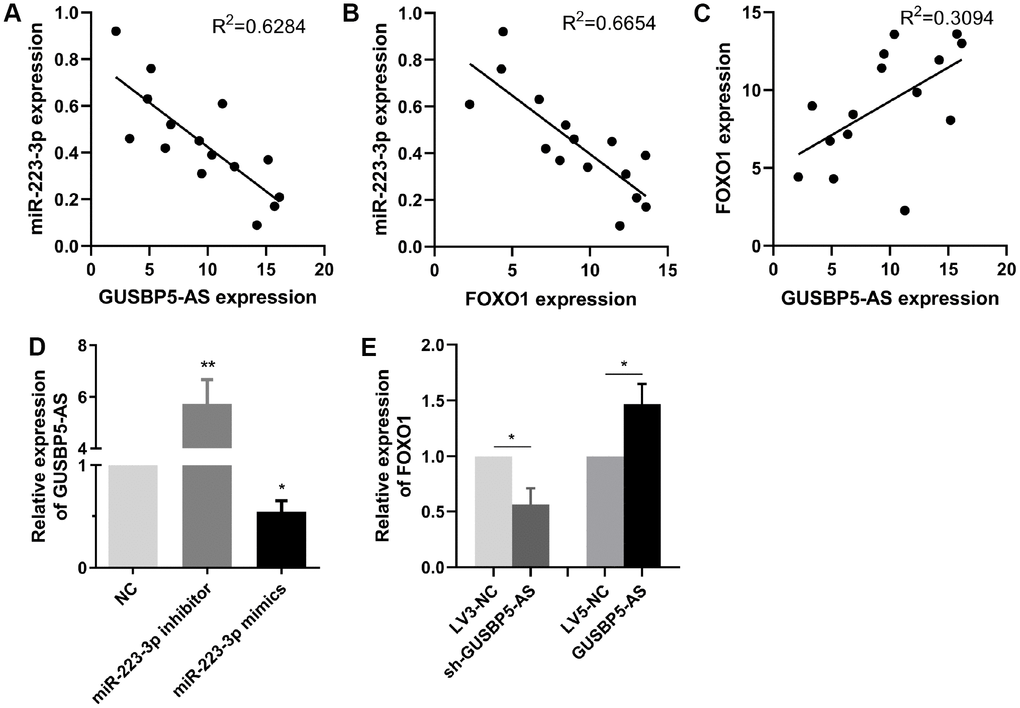
Figure 7. The relationship between GUSBP5-AS, miR-223-3p and FOXO1 expression. (A–C) Spearman rank correlation analyses showed negative correlation with GUSBP5-AS and miR-223-3p in 15 cases of EPCs samples from DVT patients, and FOXO1 was negatively correlated with miR-223-3p expression, while GUSBP5-AS was positively correlated with FOXO1 expression. (D) miR-223-3p mimics and inhibitor, respectively, inhibited and enhanced the expression of GUSBP5-AS in EPCs. (E) GUSBP5-AS knockdown and overexpression, respectively, reduced and increased the mRNA expression levels of FOXO1 in EPCs. *P < .05, **P < .01 compared to the corresponding control group. Data are represented as mean ± SEM.
Role of GUSBP5-AS and miR-223-3p in the regulation of the FOXO1/Akt pathway, FGF2 and MMP2/9 expression
To further explore the effects of GUSBP5-AS on intracellular pathways and the factors related to angiogenesis and migration, the protein expression levels of FOXO1, p-Akt, FGF2, MMP2 and MMP9 were detected by western blot, indicating that GUSBP5-AS knockdown significantly reduced these protein levels, while GUSBP5-AS overexpression increased them (Figure 8A). Importantly, our data showed that miR-223-p inhibitor enhanced FOXO1, p-Akt, FGF2, MMP2 and MMP9 expression whereas miR-223-p mimics suppressed their expression levels (Figure 8B). Interestingly, according to the previous study, FOXO1 can feed back to Akt and activate Akt pathway [36]. Thus, our results revealed that GUSBP5-AS played a central role in the regulation of angiogenesis in vivo and in vitro, migration and invasion ability of EPCs by regulating the expression levels of FGF2, MMP2 and MMP9 through miR-223-3p/FOXO1/Akt pathway.
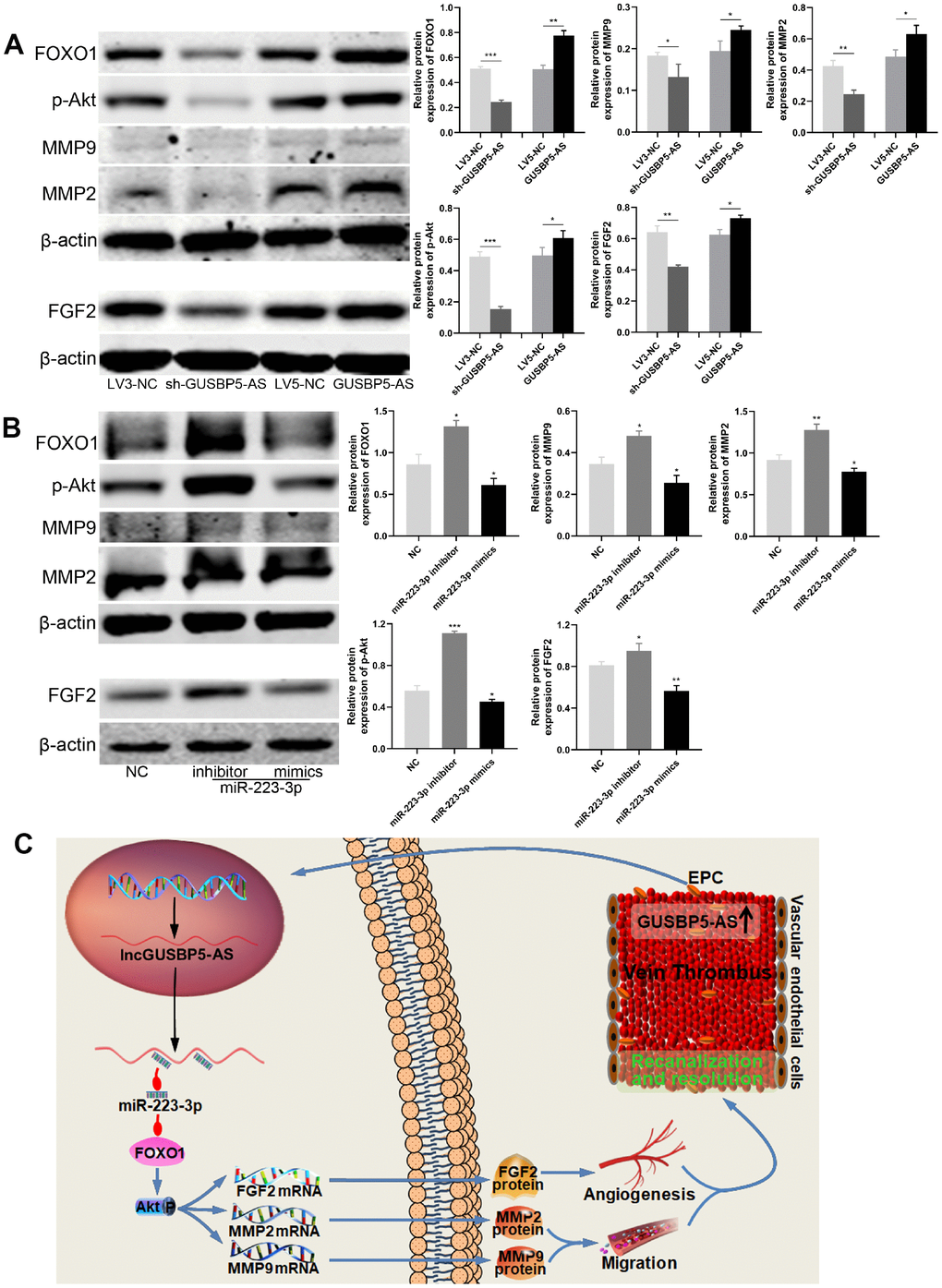
Figure 8. GUSBP5-AS regulates FGF2 and MMP2/9 expression through FOXO1/Akt pathway by sponging with miR-223-3p. (A) Effects of GUSBP5-AS on the regulation of the protein expression of FOXO1, p-Akt, MMP2, MMP9 and FGF2 in EPCs by western blot analysis. GUSBP5-AS knockdown and overexpression, respectively, reduced and increased these protein expression levels. (B) Western blot analysis of the protein expression levels of FOXO1, p-Akt, MMP2, MMP9 and FGF2 in EPCs after infected with NC, miR-223-3p inhibitor and mimics. The resulted showed that miR-223-3p inhibitor and mimics, respectively, promoted and inhibited these protein expression. *P < .05, **P < .01, ***P < .001 compared to the corresponding control group. Data are represented as mean ± SEM. (C) Schematic of the role of GUSBP5-AS-mediated effects in EPCs. Red arrow indicates inhibition and blue arrow indicates promotion.
Discussion
In this study, we demonstrated that GUSBP5-AS promotes EPC angiogenesis in vitro and in vivo and accelerates EPC homing to thrombus sites to promote DVT recanalization and resolution. In vitro, overexpression of GUSBP5-AS promoted EPC proliferation, migration, invasion and angiogenesis, and inhibited cell apoptosis. Inhibition of its expression had the opposite effects. These findings greatly extend our current understanding of the role of lncRNAs in DVT, clarifying a novel mechanism for GUSBP5-AS in EPCs and providing a new target for the management of DVT.
DVT is a common and difficult problem in the clinic. Complications such as PE and PTS caused by DVT are life-threatening [4, 5, 9]. Thrombus revascularization is an important event in the resolution of DVT [12, 13]. Thus, it is very important and necessary to develop alternative therapies to accelerate thrombus revascularization and resolution. Increasing evidence has demonstrated that EPCs have a critical role in the angiogenesis and resolution of DVT. However, the limited capacities of homing and angiogenesis impair the therapeutic effect of EPCs. Thus, it is very important to solve these limitations to improve their clinical application.
Intriguingly, recent studies have revealed that specific lncRNAs can regulate angiogenesis and development of vascular diseases [21, 37, 38]. We previously showed that GUSBP5-AS, located at chr4q31.21, is upregulated in EPCs from DVT patients compared with those from healthy controls according to microarray analysis and was predicted to be a crucial regulator in angiogenesis [32]. As shown in Figure 8C, herein we explored the effect and mechanism of GUSBP5-AS in EPCs and DVT. The results demonstrated that the up-regulated GUSBP5-AS in DVT significantly reduced thrombus size and weight, effectively accelerated EPC homing to thrombi to promote thrombus recanalization and resolution. Additionally, the mRNA-seq analysis showed GUSBP5-AS was involved in metabolic processes, stress, cell cycle, and gene expression, which is closely related to cell proliferation, migration and angiogenesis. Further, our results demonstrated that GUSBP5-AS promoted EPC proliferation, angiogenesis in vitro and in vivo, migration, invasion, F-actin filaments, FGF2 and MMP2/9 expression, and decreased apoptosis. Knockdown of GUSBP5-AS had the opposite effects. Actin and the microtubule system are required for cell adhesion and migration [39], and FGF2 and MMP2/9 are essential factors for promoting EPC migration, angiogenesis and early venous thrombus resolution [40–42]. The results suggested that GUSBP5-AS enhanced EPC proliferation, angiogenesis, migration and invasion via promoting FGF2, MMP2/9 and F-actin expression, at least in part.
MiRNAs have been demonstrated to be dysregulated in human diseases and may act as sensitive and specific biomarkers in certain vascular diseases including DVT, arteriosclerosis, aneurysm, and pulmonary arterial hypertension [43]. Growing evidence has confirmed that miRNAs have a vital role in the regulation of proliferation, migration, invasion, angiogenesis, cell cycle progression and apoptosis. MiR-223 has been shown to reduce proliferation, migration, and angiogenesis in cancer cells and endothelial cells [44, 45]. However, the role and mechanism of miR-223-3p in the regulation of EPCs and DVT has not been examined. In this study, we demonstrated the target relationships between GUSBP5-AS and miR-223-3p and between miR-223-3p and FOXO1, and we confirmed their interaction by correlation analysis. The data showed that GUSBP5-AS and FOXO1 were overexpressed in EPCs of DVT patients. Conversely, the expression level of miR-223-3p is significantly downregulated in EPCs samples of DVT patients. Moreover, our results showed that co-transfection of GUSBP5-AS and miR-223-3p further confirmed the effects of their interaction on EPC migration, invasion and tube formation, and FOXO1 knockdown and overexpression, respectively, inhibited and enhanced the abilities of angiogenesis, migration and invasion in EPCs. Together, our results suggested that the GUSBP5-AS/miR-223-3p/FOXO1 axis appears to play a crucial role in DVT progression and EPC angiogenesis, providing a novel biomarker and promising therapeutic target for DVT patients.
Notably, FGF2, a potent angiogenic factor, regulates a wide variety of functions and plays angiogenic and anti-apoptotic roles in vascular endothelial cells [46]. The matrix metalloproteinase (MMP) superfamily is composed of metalloproteinases that degrade extracellular matrix, a necessary process prior to cell migration, invasion and angiogenesis [47, 48]. Among these factors, MMP2 and MMP9 play an essential role in vascular disease [40, 49]. Previous studies have demonstrated that FGF2 and MMP2/9 can be up-regulated by activation of Akt phosphorylation [46, 47, 50]. Akt is a vital regulator of several physiological and pathological conditions, including cell proliferation, migration, angiogenesis and autophagy [50, 51]. Importantly, FOXO1, one of the most widely studied members in FOXO family, is a critical rheostat of vascular expansion, endothelial metabolism and growth [34], and deletion of Foxo1 (Foxo1 EC-KO) resulted in defective vascular development [34], and FOXO1 dependent pathways have been a research hotspot for the past decade [52]. Previous studies have found that FOXO1 can feed back to Akt and promote activation of Akt in endothelial cells [36]. In the present study, our results showed that FOXO1 was the target of miR-223-3p and promoted activation of Akt pathway, which enhanced the expression of F-actin, FGF2, MMP2 and MMP9, subsequently regulating EPC migration, angiogenesis and resulation of DVT.
In conclusion, our study substantiated that GUSBP5-AS was significantly upregulated and modulated miR-223-3p/FOXO1 axis in EPCs of DVT patients. GUSBP5-AS promotes cell angiogenesis, proliferation, migration and invasion in EPCs by regulating F-actin filaments, FGF2, MMP2 and MMP9 expression through the miR-223-3p/FOXO1/Akt pathway, thereby promoting EPC homing to thrombi and enhancing thrombosis recanalization and resolution. These findings may provide a novel biomarker and potential therapeutic target for patients with DVT.
Materials and Methods
Patients and specimens
A total of fifteen pairs of peripheral blood samples from patients with a first life-time acute DVT confirmed by ultrasonic doppler and healthy controls were obtained at The Second Affiliated Hospital of Soochow University between September 2017 and April 2018 and used to culture EPCs. None of the patients had undergone surgery or received anticoagulant drugs before blood collection. The age of DVT patients was 34-75 years, and the mean age was 54.45±15.01 years. And the age of these healthy controls was 35-72 years, and the mean age was 53.12±14.52 years. The inclusion criteria were as follows: i) Swelling, pain, and superficial varicose veins in the lower limbs and other clinical manifestations for acute DVT; and ii) DVT confirmed by ultrasound doppler. Exclusion criteria for the two groups were: i) people who had hypertension, diabetes, cardiovascular disease, liver and kidney dysfunction, or severe infection and ii) people with diseases such as immune system disease or active bleeding and other diseases. In this study, all subjects or their families signed informed consent. EPCs were identified by confocal microscopy and flow cytometry, as previously described [21].
Animals
Male nude mice were purchased from Shanghai Lingchang Biotechnology Co., Ltd. (Shanghai, China) and rasied in the Institutional Animal Care and Use Committee of Soochow University, presented in a pathogen-free facility with constant room temperature, humidity and light cycle (12:12 h light: dark), and free food and water.
Cell transfection
All oligonucleotides were purchased from GenePharma (Shanghai, China), including recombinant lentiviral particles expressing the short hairpin RNAs (shRNAs) sh-lncRNA GUSBP5-AS (targeting GUSBP5-AS), GUSBP5-AS, miR-223-3p mimics, miR-223-3p inhibitor, and their corresponding negative controls. And the sequence (5’ to 3’) of sh-GUSBP5-AS is AGCATGAACTTCACGTCAAGA. Lentiviruses were used to infect EPCs according to the manufacturer’s instructions and polybrene (5 μg/mL) was used to enhance infection efficiency. Cells were then selected by 3 μg/mL puromycin (Invivogen, San Diego, CA, USA) for 1 week to construct stable GUSBP5-AS or miR-223-3p overexpression and knockdown cell lines along with controls.
Analysis of the therapeutic effect on deep vein thrombosis
Nude mice (10 weeks old) were anesthetized with 1% pentobarbital sodium at 10 μL/g body weight. The left jugular vein of the nude mouse was isolated under a stereo microscope, and the proximal end of the heart and its branches were ligated. GFP-GUSBP5-AS EPCs and GFP-LV5-NC EPCs (5×106) were injected into the nude mice through the orbital vein. After 7 days, thrombi with vessel walls were harvested for measurements of size and weight, and for immunofluorescence. The number of GFP-EPCs at the thrombus was observed under an IX-81 laser confocal microscope (Olympus) to analyze homing ability. To immunostain CD34 and MMP2, frozen sections were stained with anti-CD34 (abcam, Cambridge, UK; ab81289, 1:100) or anti-MMP2 (abcam, ab92536, 1:200). After incubation with fluorescent-labeled secondary antibodies (donkey anti-rabbit Alexa Fluor® 647, abcam, 1:500) at room temperature for 60 min, images were observed with a confocal microscope.
In vivo angiogenesis assay
200μL Matrigel (BD), and Matrigel mixture containing EPCs (5×105) infected with GUSBP5-AS, sh-GUSBP5-AS and corresponding negative controls were implanted on the flank of the 5-week-old male nude mice. After 7 days, Matrigel implants were removed, fixed in 4% paraformaldehyde, embedded in paraffin, and then sectioned. Histological sections were stained with hematoxylin and eosin (H&E) to detect the presence of luminal structures. Images were obtained by an Olympus DP73 microscope (Tokyo, Japan). Microvascular structures were counted in three high-power views (200× magnification) of each section.
Matrigel tube formation assay
Transfected EPCs (5×104 cells) were seeded in Matrigel-coated wells of a 24-well plate to induce tube formation. After 12 h of incubation, the tubular structures of EPCs in the Matrigel were examined by microscopy, and the extent of tube formation was assessed by measuring the tube number and tube length with ImageJ software. At least five random fields (100× magnification) per well were examined to determine cell tube formation ability. All experiments were repeated three times.
Wound healing assay
Wound healing migration assays were performed with EPCs in 6-well plates. When the cells had grown to nearly 90% confluency, the cell monolayer was scratched using a 200 μL pipette tip to create a wound. After removing the cellular debris with PBS, the cells were incubated with serum-free EBM-2 medium for a specific duration. Cell migration was observed using an inverted microscope and cells were imaged at 0 and 12 h after the scratch. The wound size was quantified using Image J in three wells per group.
Transwell assay
Cell invasion assays were performed using 24-well Matrigel chambers (8-mm pore size; BD, USA) containing 12 μL Matrigel-coated membranes. Each cell suspension in serum-free EBM-2 medium was counted by microscopy. Medium containing 10% FBS (300 μL) was added to the lower wells. Each group of cells (2×104 cells in 200 μL serum-free EBM-2 medium) was added to the upper transwell chambers. Matrigel chambers were incubated at 37°C in a 5% CO2 incubator for 24 h.
Proliferation assay
Cell proliferation was measured using a Cell Counting Kit-8 (CCK-8). The transfected EPCs were seeded into 96-well plates. After 0, 24, 48, 72 and 96 h, the medium was replaced with 100 μL detection buffer (ratio of medium and CCK-8, 9:1) and cells were incubated for 2 h. The absorbance at 450 nm was detected by a microplate reader (Bio-Rad). Experiments were performed in triplicate.
Cell cycle assay
Cells were harvested by trypsinization and fixed with 70% ethanol at 4°C overnight. The fixed cells were washed with PBS and successively incubated with propidium iodide (PI; 50 μg/mL) and RNase A (100 μg/mL) for 1 h at room temperature, followed by analysis by flow cytometry. All tests were performed in triplicate.
Apoptosis assay
Cell apoptosis was analyzed using a BD Accuri™ C6 flow cytometer (BD Biosciences, USA) and an Annexin V-APC/7-AAD Apoptosis Detection Kit (KeyGEN BioTECH), according to the manufacturer’s instructions. Flow cytometric analysis of 7-AAD-Annexin V staining was repeated at least three times.
Immunofluorescence
Transfected EPCs were grown on cover slides for 24 h. Cells were washed with PBS and then fixed with 4% paraformaldehyde for 10 min at room temperature. After three washes, the cells were blocked with Immunol Staining Blocking Buffer (Beyotime, P0102) for 1 h at room temperature. Immunostaining was performed using TRITC Phalloidin (Solarbio, CA1610) to identify F-actin. Cell nuclei were stained with DAPI. The coverslips were washed with PBS and inverted onto slides that had been treated with Fluoromount-GTM seal. Images were acquired by confocal microscopy under the same conditions for each experiment, and no digital manipulation of images was performed.
RNAseq assay
Total RNA was extracted from 1×107 cells using RNAiso Plus (TaKaRa). RNAseq was performed using EPCs stably expressing empty vector (LV5-NC) or GUSBP5-AS. An RNA library was generated according to Illumina’s RNAseq protocol. The samples were fragmented and primed for cDNA synthesis, adapters were ligated to the cDNA, and the resulting samples were amplified by PCR. Sequencing was performed on Illumina HiSeq 2000 paired-end module (Illumina).
Luciferase activity assay
For luciferase reporter assays, the 3’UTR segments of FOXO1 predicted to interact with miR-223-3p were amplified by PCR. Cells were cotransfected with firefly luciferase reporter plasmid (lncRNA GUSBP5-AS-WT, lncRNA GUSBP5-AS-MU, FOXO1-WT, FOXO1-MU) and a Renilla luciferase vector (pMIR-Report Luciferase, Promega) plus negative control and small RNAs (NC, miR-223-3p mimics) using Lipofectamine 2000 (Invitrogen, USA). Experiments were performed at least three times. Cells were harvested 48 h later, and firefly and Renilla luciferase activities were analyzed with a Modulus™ single tube multimode reader (Sunnyvale, CA, USA).
Quantitative reverse transcriptase-PCR (qRT-PCR) analysis
Total RNA was extracted from harvested cells using RNAiso Plus (TaKaRa, Otsu, Japan), and first-strand cDNA synthesis was performed using a PrimeScript RT reagent kit (TaKaRa). qRT-PCR analysis was carried out using SYBR Green quantitative PCR (qPCR) Master Mix (Bio-Rad, CA, USA). MiRNA levels were measured using the Hairpin-it™. miRNAs RT-PCR Quantitation Kit (GenePharma) according to the manufacturer’s instructions. mRNA expression levels were analyzed using mRNA primers (GENEWIZ, Jiangsu, China). The endogenous control for miR-223-3p was U6 RNA and GAPDH for the others. qRT-PCR was performed on a High Throughput Quantitative PCR LightCycler480 II (Roche, Basel, Switzerland). The relative expression levels of each candidate gene were calculated using the 2^-ΔΔ CT method [24]. Each experiment was performed independently and in triplicate. The qRT-PCR primers are shown in Supplementary Table 1.
Western blot analysis
Transfected cells were cultured for 48 h. Total cellular protein was isolated with RIPA buffer (50 mM Tris-HCL pH 7.4, 150 mM NaCl, 1%NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) supplemented with protease inhibitors, phosphatase inhibitors, and PMSF. Cellular debris was removed by centrifugation at 12000 g for 10 min at 4°C. Protein quantification was performed using a BCA Protein Assay Kit (Beyotime, P0011). Lysates were separated by 10% or 15% SDS PAGE gels and transferred to PVDF membranes (Bio-Rad). Membranes were subjected to western blotting using antibodies against FOXO1 (Cell Signaling, 1:1000), p-Akt (Cell Signaling, 1:1000), FGF2 (Cell Signaling, 1:1000), MMP2 (Abcam, 1:1000), MMP9 (Abcam, 1:1000) and β-actin (Cell Signaling, 1:1000). IRDye 800CW goat anti-rabbit or anti-mouse IgG (Licor) was used at 1:10,000 dilution. Infrared Imagine System (LI-COR) was performed to analyze proteins.
Statistical analysis
In all in vitro studies, three separate experiments were performed on cells from different transfection experiments. Data are presented as mean ± standard error of the mean (SEM). Student’s t-test was used for comparing means between two groups. Statistical significance was analyzed by one-way ANOVA in three or more groups using GraphPad Prism 8 software. P-value of 0.05 or less was considered statistically significant.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
Funding
This study was sponsored by grants from the National Natural Science Foundation of China (No. 81770483, 81800418), the Natural Science Foundation of Jiangsu Province (No. BK20180125), the Key Project supported by Medical Science and Technology Development Foundation (No. YKK18063, ZKX18021), Postgraduate Research and Practice Innovation Program of Jiangsu Province (No. KYCX19-1985).
References
- 1. Cushman M. Epidemiology and risk factors for venous thrombosis. Semin Hematol. 2007; 44:62–69. https://doi.org/10.1053/j.seminhematol.2007.02.004 [PubMed]
- 2. Turetz M, Sideris AT, Friedman OA, Triphathi N, Horowitz JM. Epidemiology, Pathophysiology, and Natural History of Pulmonary Embolism. Semin Intervent Radiol. 2018; 35:92–98. https://doi.org/10.1055/s-0038-1642036 [PubMed]
- 3. Heit JA, Mohr DN, Silverstein MD, Petterson TM, O’Fallon WM, Melton LJ
3rd . Predictors of recurrence after deep vein thrombosis and pulmonary embolism: a population-based cohort study. Arch Intern Med. 2000; 160:761–68. https://doi.org/10.1001/archinte.160.6.761 [PubMed] - 4. Giordano NJ, Jansson PS, Young MN, Hagan KA, Kabrhel C. Epidemiology, Pathophysiology, Stratification, and Natural History of Pulmonary Embolism. Tech Vasc Interv Radiol. 2017; 20:135–40. https://doi.org/10.1053/j.tvir.2017.07.002 [PubMed]
- 5. Boon GJ, Van Dam LF, Klok FA, Huisman MV. Management and treatment of deep vein thrombosis in special populations. Expert Rev Hematol. 2018; 11:685–95. https://doi.org/10.1080/17474086.2018.1502082 [PubMed]
- 6. Tapson VF, Humbert M. Incidence and prevalence of chronic thromboembolic pulmonary hypertension: from acute to chronic pulmonary embolism. Proc Am Thorac Soc. 2006; 3:564–67. https://doi.org/10.1513/pats.200605-112LR [PubMed]
- 7. Yago T, Liu Z, Ahamed J, McEver RP. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. 2018; 132:1426–37. https://doi.org/10.1182/blood-2018-05-850859 [PubMed]
- 8. Prandoni P, Lensing AW, Cogo A, Cuppini S, Villalta S, Carta M, Cattelan AM, Polistena P, Bernardi E, Prins MH. The long-term clinical course of acute deep venous thrombosis. Ann Intern Med. 1996; 125:1–7. https://doi.org/10.7326/0003-4819-125-1-199607010-00001 [PubMed]
- 9. Ueda J, Tsuji A, Ogo T, Asano R, Konagai N, Fukui S, Morita Y, Fukuda T, Yasuda S. Beneficial Effect of Endovascular Treatment on Villalta Score in Japanese Patients With Chronic Iliofemoral Venous Thrombosis and Post-Thrombotic Syndrome. Circ J. 2018; 82:2640–46. https://doi.org/10.1253/circj.CJ-17-1210 [PubMed]
- 10. Diaz JA, Ramacciotti E, Wakefield TW. Do galectins play a role in venous thrombosis? a review. Thromb Res. 2010; 125:373–76. https://doi.org/10.1016/j.thromres.2009.11.011 [PubMed]
- 11. Heit JA, Cohen AT, Anderson FA. Estimated annual number of incident and recurrent, non-fatal and fatal venous thromboembolism (VTE) events in the US. Blood. 2005; 106:267A.
- 12. Sun J, Zhang Z, Ma T, Yang Z, Zhang J, Liu X, Lu D, Shen Z, Yang J, Meng Q. Endothelial progenitor cell-derived exosomes, loaded with miR-126, promoted deep vein thrombosis resolution and recanalization. Stem Cell Res Ther. 2018; 9:223. https://doi.org/10.1186/s13287-018-0952-8 [PubMed]
- 13. Santo SD, Tepper OM, von Ballmoos MW, Diehm N, Völzmann J, Baumgartner I, Kalka C. Cell-based therapy facilitates venous thrombus resolution. Thromb Haemost. 2009; 101:460–64. https://doi.org/10.1160/TH08-07-0453 [PubMed]
- 14. Modarai B, Burnand KG, Sawyer B, Smith A. Endothelial progenitor cells are recruited into resolving venous thrombi. Circulation. 2005; 111:2645–53. https://doi.org/10.1161/CIRCULATIONAHA.104.492678 [PubMed]
- 15. Modarai B, Burnand KG, Humphries J, Waltham M, Smith A. The role of neovascularisation in the resolution of venous thrombus. Thromb Haemost. 2005; 93:801–09. https://doi.org/10.1160/TH04-09-0596 [PubMed]
- 16. Mo J, Zhang D, Yang R. MicroRNA-195 regulates proliferation, migration, angiogenesis and autophagy of endothelial progenitor cells by targeting GABARAPL1. Biosci Rep. 2016; 36:e00396. https://doi.org/10.1042/BSR20160139 [PubMed]
- 17. Miller-Kasprzak E, Jagodziński PP. Endothelial progenitor cells as a new agent contributing to vascular repair. Arch Immunol Ther Exp (Warsz). 2007; 55:247–59. https://doi.org/10.1007/s00005-007-0027-5 [PubMed]
- 18. Werner N, Nickenig G. Influence of cardiovascular risk factors on endothelial progenitor cells: limitations for therapy? Arterioscler Thromb Vasc Biol. 2006; 26:257–66. https://doi.org/10.1161/01.ATV.0000198239.41189.5d [PubMed]
- 19. Al Mheid I, Hayek SS, Ko YA, Akbik F, Li Q, Ghasemzadeh N, Martin GS, Long Q, Hammadah M, Maziar Zafari A, Vaccarino V, Waller EK, Quyyumi AA. Age and Human Regenerative Capacity Impact of Cardiovascular Risk Factors. Circ Res. 2016; 119:801–09. https://doi.org/10.1161/CIRCRESAHA.116.308461 [PubMed]
- 20. Hill JM, Zalos G, Halcox JP, Schenke WH, Waclawiw MA, Quyyumi AA, Finkel T. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003; 348:593–600. https://doi.org/10.1056/NEJMoa022287 [PubMed]
- 21. Li WD, Zhou DM, Sun LL, Xiao L, Liu Z, Zhou M, Wang WB, Li XQ. LncRNA WTAPP1 Promotes Migration and Angiogenesis of Endothelial Progenitor Cells via MMP1 Through MicroRNA 3120 and Akt/PI3K/Autophagy Pathways. Stem Cells. 2018; 36:1863–74. https://doi.org/10.1002/stem.2904 [PubMed]
- 22. Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Mol Cell. 2011; 43:904–14. https://doi.org/10.1016/j.molcel.2011.08.018 [PubMed]
- 23. Weng R, Lu C, Liu X, Li G, Lan Y, Qiao J, Bai M, Wang Z, Guo X, Ye D, Jiapaer Z, Yang Y, Xia C, et al. Long Noncoding RNA-1604 Orchestrates Neural Differentiation through the miR-200c/ZEB Axis. Stem Cells. 2018; 36:325–36. https://doi.org/10.1002/stem.2749 [PubMed]
- 24. Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009; 11:228–34. https://doi.org/10.1038/ncb0309-228 [PubMed]
- 25. Uchida S, Dimmeler S. Long noncoding RNAs in cardiovascular diseases. Circ Res. 2015; 116:737–50. https://doi.org/10.1161/CIRCRESAHA.116.302521 [PubMed]
- 26. Leone S, Santoro R. Challenges in the analysis of long noncoding RNA functionality. FEBS Lett. 2016; 590:2342–53. https://doi.org/10.1002/1873-3468.12308 [PubMed]
- 27. Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, Ponnusamy M, Shan C, Xu S, et al. LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription. Nat Commun. 2018; 9:29. https://doi.org/10.1038/s41467-017-02280-y [PubMed]
- 28. Li N, Ponnusamy M, Li MP, Wang K, Li PF. The Role of MicroRNA and LncRNA-MicroRNA Interactions in Regulating Ischemic Heart Disease. J Cardiovasc Pharmacol Ther. 2017; 22:105–11. https://doi.org/10.1177/1074248416667600 [PubMed]
- 29. Ouseph MM, Huang Y, Banerjee M, Joshi S, MacDonald L, Zhong Y, Liu H, Li X, Xiang B, Zhang G, Komatsu M, Yue Z, Li Z, et al. Autophagy is induced upon platelet activation and is essential for hemostasis and thrombosis. Blood. 2015; 126:1224–33. https://doi.org/10.1182/blood-2014-09-598722 [PubMed]
- 30. Zhao H, Diao C, Wang X, Xie Y, Liu Y, Gao X, Han J, Li S. LncRNA BDNF-AS inhibits proliferation, migration, invasion and EMT in oesophageal cancer cells by targeting miR-214. J Cell Mol Med. 2018; 22:3729–39. https://doi.org/10.1111/jcmm.13558 [PubMed]
- 31. Li X, Wu Z, Fu X, Han W. Long Noncoding RNAs: Insights from Biological Features and Functions to Diseases. Med Res Rev. 2013; 33:517–53. https://doi.org/10.1002/med.21254 [PubMed]
- 32. Qian AM, Li WD, Kong LS, Rong J, Lei FR, Du XL, Li CL, Li XQ. Long noncoding RNA expression profile of endothelial progenitor cells from deep vein thrombosis patients identified by microarray analysis. Int J Clin Exp Pathol. 2016; 9:12275–86.
- 33. Li WD, Li XQ. Endothelial progenitor cells accelerate the resolution of deep vein thrombosis. Vascul Pharmacol. 2016; 83:10–16. https://doi.org/10.1016/j.vph.2015.07.007 [PubMed]
- 34. Wilhelm K, Happel K, Eelen G, Schoors S, Oellerich MF, Lim R, Zimmermann B, Aspalter IM, Franco CA, Boettger T, Braun T, Fruttiger M, Rajewsky K, et al. FOXO1 couples metabolic activity and growth state in the vascular endothelium. Nature. 2016; 529:216–20. https://doi.org/10.1038/nature16498 [PubMed]
- 35. Jeon HH, Yu Q, Lu Y, Spencer E, Lu C, Milovanova T, Yang Y, Zhang C, Stepanchenko O, Vafa RP, Coelho PG, Graves DT. FOXO1 regulates VEGFA expression and promotes angiogenesis in healing wounds. J Pathol. 2018; 245:258–64. https://doi.org/10.1002/path.5075 [PubMed]
- 36. Dharaneeswaran H, Abid MR, Yuan L, Dupuis D, Beeler D, Spokes KC, Janes L, Sciuto T, Kang PM, Jaminet SS, Dvorak A, Grant MA, Regan ER, Aird WC. FOXO1-mediated activation of Akt plays a critical role in vascular homeostasis. Circ Res. 2014; 115:238–51. https://doi.org/10.1161/CIRCRESAHA.115.303227 [PubMed]
- 37. Chen L, Yang W, Guo Y, Chen W, Zheng P, Zeng J, Tong W, Tong W. Exosomal lncRNA GAS5 regulates the apoptosis of macrophages and vascular endothelial cells in atherosclerosis. PLoS One. 2017; 12:e0185406. https://doi.org/10.1371/journal.pone.0185406 [PubMed]
- 38. Lino Cardenas CL, Kessinger CW, Cheng Y, MacDonald C, MacGillivray T, Ghoshhajra B, Huleihel L, Nuri S, Yeri AS, Jaffer FA, Kaminski N, Ellinor P, Weintraub NL, et al. An HDAC9-MALAT1-BRG1 complex mediates smooth muscle dysfunction in thoracic aortic aneurysm. Nat Commun. 2018; 9:1009. https://doi.org/10.1038/s41467-018-03394-7 [PubMed]
- 39. Wehrle-Haller B, Imhof BA. Actin, microtubules and focal adhesion dynamics during cell migration. Int J Biochem Cell Biol. 2003; 35:39–50. https://doi.org/10.1016/S1357-2725(02)00071-7 [PubMed]
- 40. Sood V, Luke CE, Deatrick KB, Baldwin J, Miller EM, Elfline M, Upchurch GR
Jr , Wakefield TW, Henke PK. Urokinase plasminogen activator independent early experimental thrombus resolution: MMP2 as an alternative mechanism. Thromb Haemost. 2010; 104:1174–83. https://doi.org/10.1160/TH10-03-0184 [PubMed] - 41. Visse R, Nagase H. Matrix metalloproteinases and tissue inhibitors of metalloproteinases: structure, function, and biochemistry. Circ Res. 2003; 92:827–39. https://doi.org/10.1161/01.RES.0000070112.80711.3D [PubMed]
- 42. Li WD, Hu N, Lei FR, Wei S, Rong JJ, Zhuang H, Li XQ. Autophagy inhibits endothelial progenitor cells migration via the regulation of MMP2, MMP9 and uPA under normoxia condition. Biochem Biophys Res Commun. 2015; 466:376–80. https://doi.org/10.1016/j.bbrc.2015.09.031 [PubMed]
- 43. Sun LL, Li WD, Lei FR, Li XQ. The regulatory role of microRNAs in angiogenesis-related diseases. J Cell Mol Med. 2018; 22:4568–87. https://doi.org/10.1111/jcmm.13700 [PubMed]
- 44. Ding Q, Shen L, Nie X, Lu B, Pan X, Su Z, Yan A, Yan R, Zhou Y, Li L, Xu J. MiR-223-3p overexpression inhibits cell proliferation and migration by regulating inflammation-associated cytokines in glioblastomas. Pathol Res Pract. 2018; 214:1330–39. https://doi.org/10.1016/j.prp.2018.05.012 [PubMed]
- 45. Shi L, Fisslthaler B, Zippel N, Frömel T, Hu J, Elgheznawy A, Heide H, Popp R, Fleming I. MicroRNA-223 antagonizes angiogenesis by targeting β1 integrin and preventing growth factor signaling in endothelial cells. Circ Res. 2013; 113:1320–30. https://doi.org/10.1161/CIRCRESAHA.113.301824 [PubMed]
- 46. Yang TC, Chen YJ, Chang SF, Chen CH, Chang PY, Lu SC. Malondialdehyde mediates oxidized LDL-induced coronary toxicity through the Akt-FGF2 pathway via DNA methylation. J Biomed Sci. 2014; 21:11. https://doi.org/10.1186/1423-0127-21-11 [PubMed]
- 47. Sze KM, Wong KL, Chu GK, Lee JM, Yau TO, Ng IO. Loss of phosphatase and tensin homolog enhances cell invasion and migration through AKT/Sp-1 transcription factor/matrix metalloproteinase 2 activation in hepatocellular carcinoma and has clinicopathologic significance. Hepatology. 2011; 53:1558–69. https://doi.org/10.1002/hep.24232 [PubMed]
- 48. Heissig B, Hattori K, Dias S, Friedrich M, Ferris B, Hackett NR, Crystal RG, Besmer P, Lyden D, Moore MA, Werb Z, Rafii S. Recruitment of stem and progenitor cells from the bone marrow niche requires MMP-9 mediated release of kit-ligand. Cell. 2002; 109:625–37. https://doi.org/10.1016/S0092-8674(02)00754-7 [PubMed]
- 49. Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res. 2002; 90:251–62. https://doi.org/10.1161/res.90.3.251 [PubMed]
- 50. Liu T, Zhou W, Cai B, Chu J, Shi G, Teng H, Xu J, Xiao J, Wang Y. IRX2-mediated upregulation of MMP-9 and VEGF in a PI3K/AKT-dependent manner. Mol Med Rep. 2015; 12:4346–51. https://doi.org/10.3892/mmr.2015.3915 [PubMed]
- 51. Tao Z, Feng C, Mao C, Ren J, Tai Y, Guo H, Pu M, Zhou Y, Wang G, Wang M. MiR-4465 directly targets PTEN to inhibit AKT/mTOR pathway-mediated autophagy. Cell Stress Chaperones. 2019; 24:105–13. https://doi.org/10.1007/s12192-018-0946-6 [PubMed]
- 52. Huang Y, Fan X, Tao R, Song Q, Wang L, Zhang H, Kong H, Huang J. Effect of miR-182 on hepatic fibrosis induced by Schistosomiasis japonica by targeting FOXO1 through PI3K/AKT signaling pathway. J Cell Physiol. 2018; 233:6693–704. https://doi.org/10.1002/jcp.26469 [PubMed]