



Introduction
Aging is an independent risk factor for cardiovascular disorders including ischemic heart diseases [1]. Aged hearts are more likely to fail in ischemia-reperfusion injury (I/R) compared to younger hearts, as endogenous antioxidative capacity declines with aging. Aging is also associated with oxidative protein modifications and loss of function [2]. However, interventions with antioxidants have not been very effective against I/R injury suggesting that additional mechanisms are involved in high incidence of age-dependent cardiovascular diseases. In this regard it is established that reactive oxygen species (ROS), which are produced during the reperfusion of the myocardium cause extensive damage to the affected heart tissue [3]. Although uncoupled endothelial nitric oxide synthase (eNOS), xanthine oxidase (XO), NADPH oxidases (Nox) and mitochondria are implicated as sources of ROS, mitochondria-mediated ROS release is of critical importance in cardiomyocyte death via apoptosis or necrosis [4–6].
We have previously demonstrated that aged heart mitochondria respire at a lower rate compared to young mice, and metabolic demand on oxidation of mitochondrial fuels are decreased with age [7]. The energy mitochondria generate by oxidative phosphorylation is required for normal heart function. Paradoxically, mitochondria are also significant source of the ROS generated during normal respiration [8]. Altered mitochondrial structure and function in the aging process have been shown to aggravate I/R-mediated mitochondrial dysfunction [9]. Age-related defects in various mitochondrial ETC complexes combined with increased mitochondrial fission and decreased fusion process impairs overall mitochondrial capacity in the aging, which is further deteriorated during I/R [10, 11]. Mitochondrial biogenesis led by peroxisome proliferator-activated receptor γ (PPARγ) coactivator-1α (PGC1α), is an important member of transcriptional coactivators family [12], regulates mitochondrial energy metabolism and cardiac function [13]. PGC1α is regulated by transcription and posttranslational modifications, such as phosphorylation, acetylation and methylation. In addition to promoting mitochondrial biogenesis, PGC1α has also been shown to be involved in the induction of several ROS detoxifying enzymes [14]. Ectopic expression of PGC1α in muscle cells induces superoxide dismutase-2 and glutathione peroxidase I, both of which are involved in removal of ROS.
Trx is a small (12 kDa) redox protein that is an electron donor for ribonucleotide reductase for the synthesis of deoxyribonucleotides, a rate-limiting step in DNA replication [15]. Trx is also an electron donor for peroxiredoxins, which detoxify peroxides [16]. Thioredoxin reductase-1 (TrxR1) uses reducing equivalents form NADPH and transfers electrons to recycle oxidized Trx produced in redox reactions to reduce Trx [17]. Trx scavenges hydroxyl radicals, quenches singlet oxygen, and induces mitochondrial SOD2 [18, 19]. A mitochondrial thioredoxin-2 (Trx-2) is present in the mitochondria of cells. Although active center cysteines of Trx is preserved in Trx-2, it lacks the additional structural cysteines that are present in Trx. Trx-2 has been shown to diminish mitochondrial ROS [20], and decreases myocardial apoptosis by reducing mitochondrial ROS [21]. We have previously shown that Trx regulates MAP Kinase Kinase-4 (MKK4) activation via redox regulation resulting in sequential activation of NFκB and AP-1, which regulates SOD2 expression in endothelial cells [19]. In addition, a major function of Trx includes regeneration of –SH group enzymes and proteins, which are inactivated by oxidation [15]. Thus, Trx not only is a radical scavenger or inducer of SOD2, but also converts oxidized proteins to their native state through its disulfide reductase properties. We have earlier demonstrated that high levels of Trx prevent I/R injury in adult mouse heart by eNOS deglutathionylation [22] and prevent age –related hypertension involving vascular mechanisms in mice [23]. Additionally, a previous study has demonstrated decreased post-myocardial apoptosis by recombinant human Trx [24]. Since I/R injury is a multi-factorial and results from oxidative protein modifications, mitochondrial dysfunction and alteration of redox state, we hypothesized that Trx would ameliorate I/R injury via regenerating oxidized proteins to their native state in the face of I/R –mediated modifications, and due to preservation of redox state in aged mice.
To determine specific redox-related mechanisms by which Trx ameliorates I/R injury in aging, we utilized Trx-Tg and dnTrx-Tg mice [25]. The dnTrx-Tg mice maintain only low amounts of active Trx (3-5 fold lower) because of a dominant-negative effect of the mutant protein in preventing redox-related actions of Trx via competitive inhibition for reduction by TrxR1 [25]. We show that overexpression of Trx in mice protects against I/R injury by preserving myocardial redox balance, protecting mitochondrial structure and function, improving mitochondrial biogenesis by maintaining PGC1α expression and rescue of ACO2, MFN1 and MFN2 expression in I/R via AKT-CREB pathway.
Results
High levels of Trx in aged mice heart prevents I/R mediated redox shift
We determined the Trx redox state in aged mice heart and the effect of I/R to delineate how Trx may modulate myocardial redox in I/R. As shown in Figure 1A, 1B, the activities of Trx and TrxR1 are significantly lower in sham myocardium from dnTrx-Tg mice in contrast to those from NT or Trx-Tg mice, demonstrating that dnTrx-Tg mice have significantly decreased level of redox-active Trx and TrxR1 in the myocardium. Further, I/R caused marked reduction in Trx and TrxR1 activity in the infarcted region of NT mice (Figure 1A and B). However, infarcted myocardium from Trx-Tg mice showed higher Trx and TrxR1 activities compared to NT or dnTrx-Tg mice. I/R did not alter the Trx and TrxR activities further in dnTrx-Tg mice compared to respective sham animals (Figure 1A, 1B). We reasoned that oxidized Trx might have been accumulated in dnTrx-Tg mice heart in I/R due to oxidation of endogenous Trx. As shown in Figure 1C, Trx remained in oxidized state in sham or I/R treated dnTrx-Tg mice heart. However, Trx redox state in the infarcted myocardium from Trx-Tg mice was predominantly reduced, demonstrating that overexpression of Trx preserves overall redox state of the myocardium in I/R. Due to very low levels of endogenous Trx in mice, we ran separate western analysis (with higher amounts of protein) of aged sham NT or I/R subjected mice. As shown in Figure 1C (left panel), aged NT-I/R mice showed high level of oxidized Trx compared to sham treated mice. However, over expression of Trx or dnTrx did not affect Trx-2 levels (Figure 1D, 1E). Taken together, our data show that 3-fold higher active Trx in mice from the beginning of life preserves Trx redox in reduced state that protected against I/R -mediated Trx oxidation.
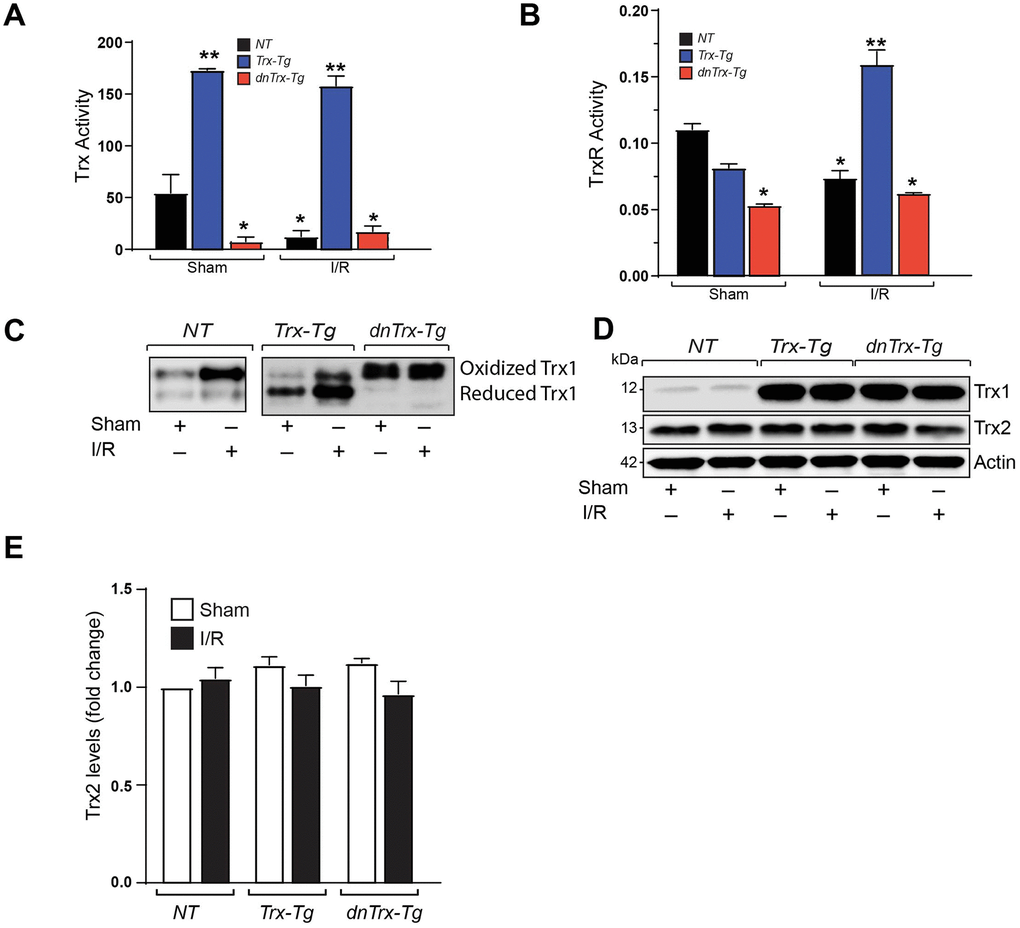
Figure 1. High amounts of hTrx in transgenic mice prevents I/R mediated redox shift, and loss of Trx and Trx reductase activities. (A) Trx activity was assayed in myocardium derived from sham and I/R-subjected NT, Trx-Tg, and dnTrx-Tg mice and expressed as nanomoles of NADPH oxidized per minute per milligram of protein at 25°C. Values are represented as means ± SEM (n =3-4). *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg. (B) TrxR activity in sham or I/R myocardium were expressed as micromoles of 5-thio-2-nitrobenzene (TNB) formed per minute per milligram of protein at 30°C. Values are represented as means ± SEM (n =3-4). *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R. (C) Redox Western blot analysis revealing the redox state of Trx (oxidized and reduced) in sham or I/R myocardium from NT, Trx-Tg and dnTrx-Tg mice. (D) AAR region of sham or I/R myocardium from NT, Trx-Tg and dnTrx-Tg were lysed using M-PER lysis buffer and analyzed for Trx1, Trx2 and Actin by western blotting. (E) Trx2 levels were quantified and expressed as fold change. Statistical significance was determined with one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test.
High levels of Trx protect against I/R-mediated LV dysfunction, reduce infarct size and decrease the expression of apoptotic proteins in aged heart
We next evaluated the effect of Trx on cardiac function in I/R. We performed echocardiography on aged NT, Trx-Tg and dnTrx-Tg mice after 60 minutes of ischemia and 30 minutes of reperfusion. As shown in Figure 2A, 2B, I/R decreased the LV ejection fraction (EF) in NT mice compared to sham animals. In contrast, Trx-Tg mice were significantly protected from I/R -mediated reduction in EF compared to NT or dnTrx-Tg mice. NT and dnTrx-Tg mice also exhibited loss of fractional shortening (FS) during I/R compared to sham animals (Figure 2C). However, Trx-Tg mice showed significant improvement in EF compared to NT or dnTrx-Tg mice subjected to I/R (Figure 2C). Next, we determined the effect of high levels of Trx on infarct size by TTC staining. As shown in Figure 2D, 2E, aged Trx-Tg mice showed significant reduction of infarct size in I/R compared to NT mice. Lower levels of active Trx in dnTrx-Tg mice accentuated the I/R insult, as evidenced by significantly higher infarct size in dnTrx-Tg mice compared to NT or Trx-Tg (Figure 2E).
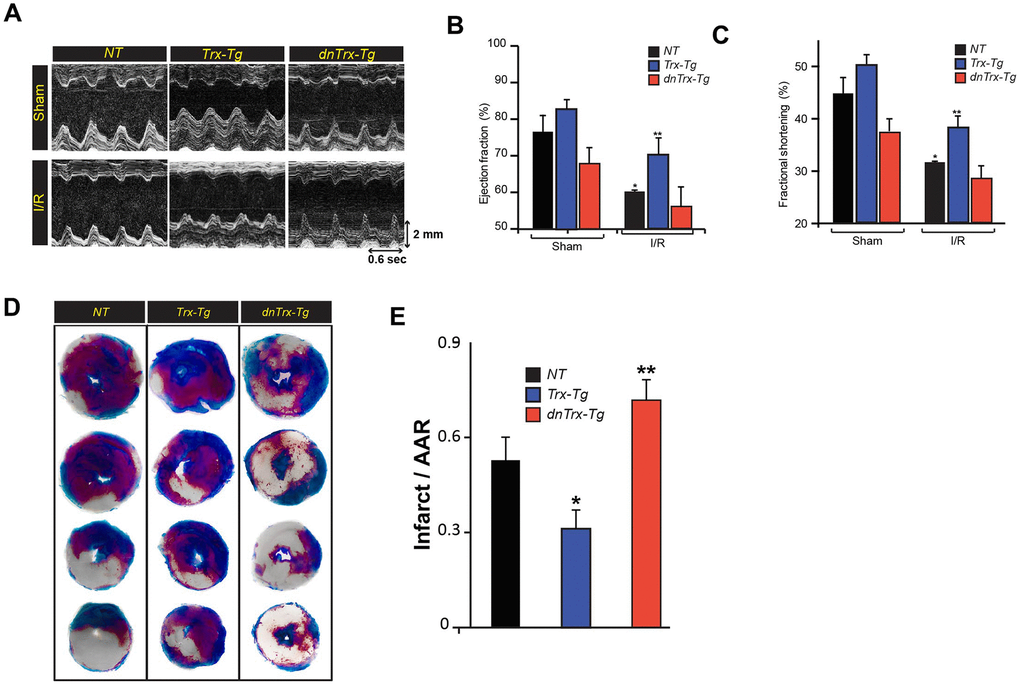
Figure 2. High level of Trx protects against I/R mediated LV dysfunction and infarction of aged myocardium. (A). Representative M-mode images taken from NT, Trx-Tg and dnTrx-Tg hearts of sham (top) and I/R (bottom). Ejection fraction; EF (B) and fractional shortening; FS (C), an index of cardiac contractile function, was determined by echocardiographic analysis. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R hearts, n=3-5. (D) NT, Trx-Tg and dnTrx-Tg mice were subjected to 60 min ischemia and 30 min reperfusion and then TTC staining was performed as described in the methods section. TTC stains viable tissue brick red and necrotic tissue as white. (E) Infarct area in relation to area-at-risk (AAR). *p <0.05 versus NT or dnTrx-Tg I/R; **p <0.05 versus NT or Trx-Tg I/R, n=4. Statistical significance was determined with the Student’s t-test.
Since apoptosis is a major contributing factor in I/R-induced death of myocytes resulting in MI, we analyzed the apoptotic markers such as release of cytochrome-c in cytosolic extracts from AAR region of sham or I/R subjected mice hearts. As shown in Figure 3A, 3B, I/R resulted in significant increase in cytochrome-c release in NT mice, however, the increase in cytochrome-c in dnTrx-Tg mice in I/R was 2-fold higher compared to NT mice. In contrast, there was no increase in cytochrome-c release in Trx-Tg mice subjected to I/R (Figure 3A, 3B). Next, we analyzed the level of cytochrome-c effector proteins, such as cleaved caspase -3 and proapoptotic Bax levels in sham or I/R to delineate the specific apoptotic pathway. As shown in Figure 3C, 3D, I/R resulted in elevated levels of cleaved caspase 3 and Bax. However, cleaved caspase levels were diminished in Trx-Tg mice in I/R. We performed an EPR apoptosis assay to determine the apoptosis in the entire infarcted region by removing the total heart tissue irrigated by LAD. As shown in Figure 3E, 3F, we found significant increase in iron-bound annexin-V in I/R in NT mice heart. In addition, the magnitude of apoptosis was further increased in dnTrx-Tg mice in I/R. Trx-Tg mice in I/R demonstrated significant decrease in myocardial apoptosis. Taken together, these data show that 3-fold increase in functional Trx in the aged myocardium protected against I/R -mediated LV dysfunction, decreased MI and reduced apoptosis. In contrast, loss of redox active Trx in the dnTrx-Tg mice exaggerated I/R injury with severe MI and apoptosis in the aged myocardium.
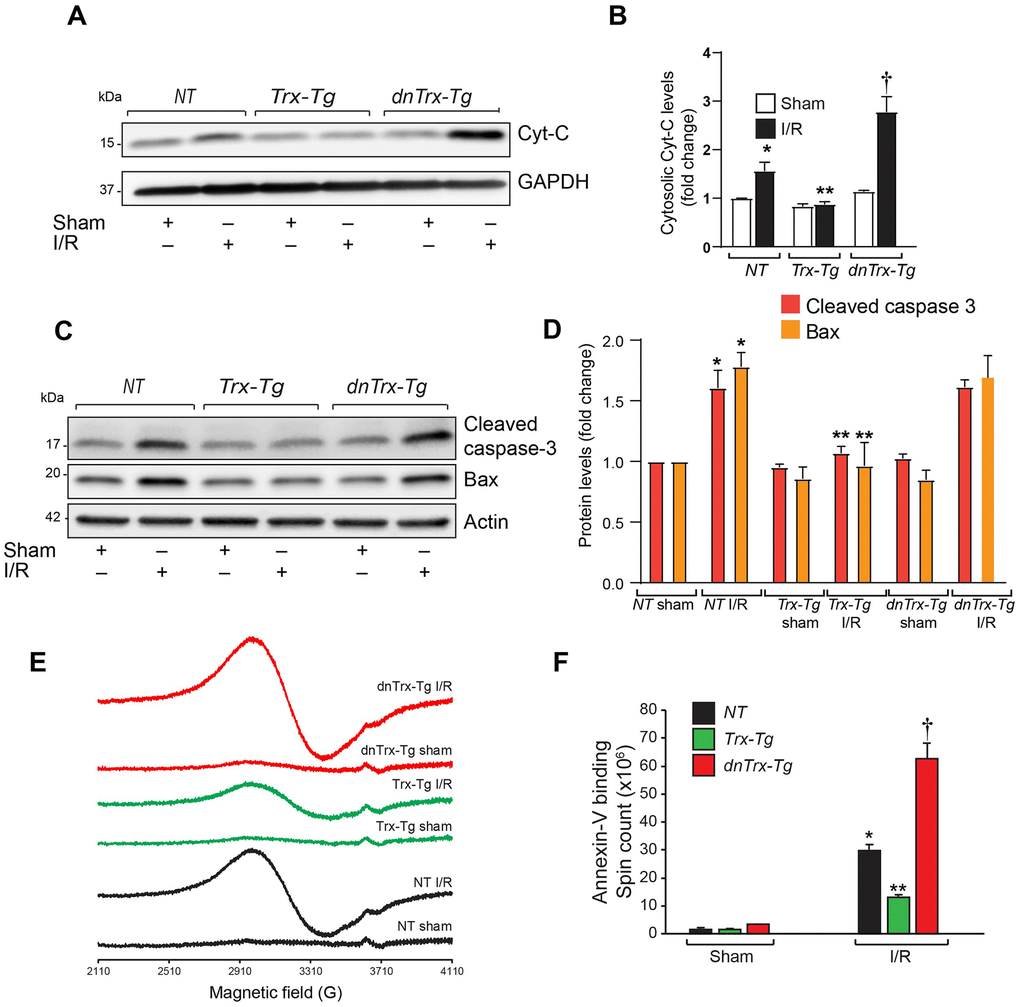
Figure 3. High level of Trx protects against I/R- induced apoptosis. (A) Cytosolic extract was prepared from AAR region of sham or I/R subjected NT, Trx-Tg and dnTrx-Tg mice and level of Cyt-C was analyzed by western blotting. (B) Levels of cytosolic Cyt-C was quantified and expressed as fold change. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R; † p <0.05 versus NT or Trx-Tg I/R. (C) AAR region of sham or I/R myocardium from NT, Trx-Tg and dnTrx-Tg were lysed using M-PER lysis buffer and analyzed for cleaved caspase 3 and Bax by western blotting. (D) Protein levels were quantified and expressed as fold change. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R hearts, n=3. (E) EPR spectra of paramagnetic iron bound Annexin-V. (F) Graph shows an absolute spin count of Fe-Annexin-V. Values are means ± SD (n = 3 mice). *, p < 0.01 versus NT Sham; **, p < 0.01 versus NT I/R or dnTrx-Tg I/R; † p <0.01 versus NT or Trx-Tg I/R. Statistical significance was determined with the Student’s t test (B, D) and one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test (F).
High levels of Trx protect cardiac mitochondrial structure, function and prevents mitochondrial DNA damage due to I/R in aged mice
Since we found decreased levels of cytochrome-c, bax and cleaved caspase-3 in Trx-Tg mice in I/R, which are indicative of mitochondrial pathway of apoptosis, we reasoned that mitochondrial dysfunction might have been impacted by high levels of Trx in the hearts of Trx-Tg mice in I/R. Therefore, we determined the effect of high levels of Trx on mitochondrial function, mitochondrial ultrastructure, mitochondrial DNA damage and mitochondrial enzyme activities in sham and I/R animals. We analyzed the ultrastructure of myocardium by electron microscopy. As shown in Figure 4A, I/R caused significant loss of cristae in NT mice (upper left panels) and dnTrx-Tg mice in I/R (lower left panels). In contrast, the mitochondrial structure was preserved in Trx-Tg mice that showed normal cristae structure and density in I/R (top right panels; andFigure 4B). Additionally, as shown in Figure 4C, I/R caused significant increase in damaged mitochondria in aged NT or dnTrx-Tg mice, but not in aged Trx-Tg mice in I/R. Further, mitochondrial swelling was increased in I/R in aged NT or dnTrx-Tg mice, but not in aged Trx-Tg mice (Figure 4D).
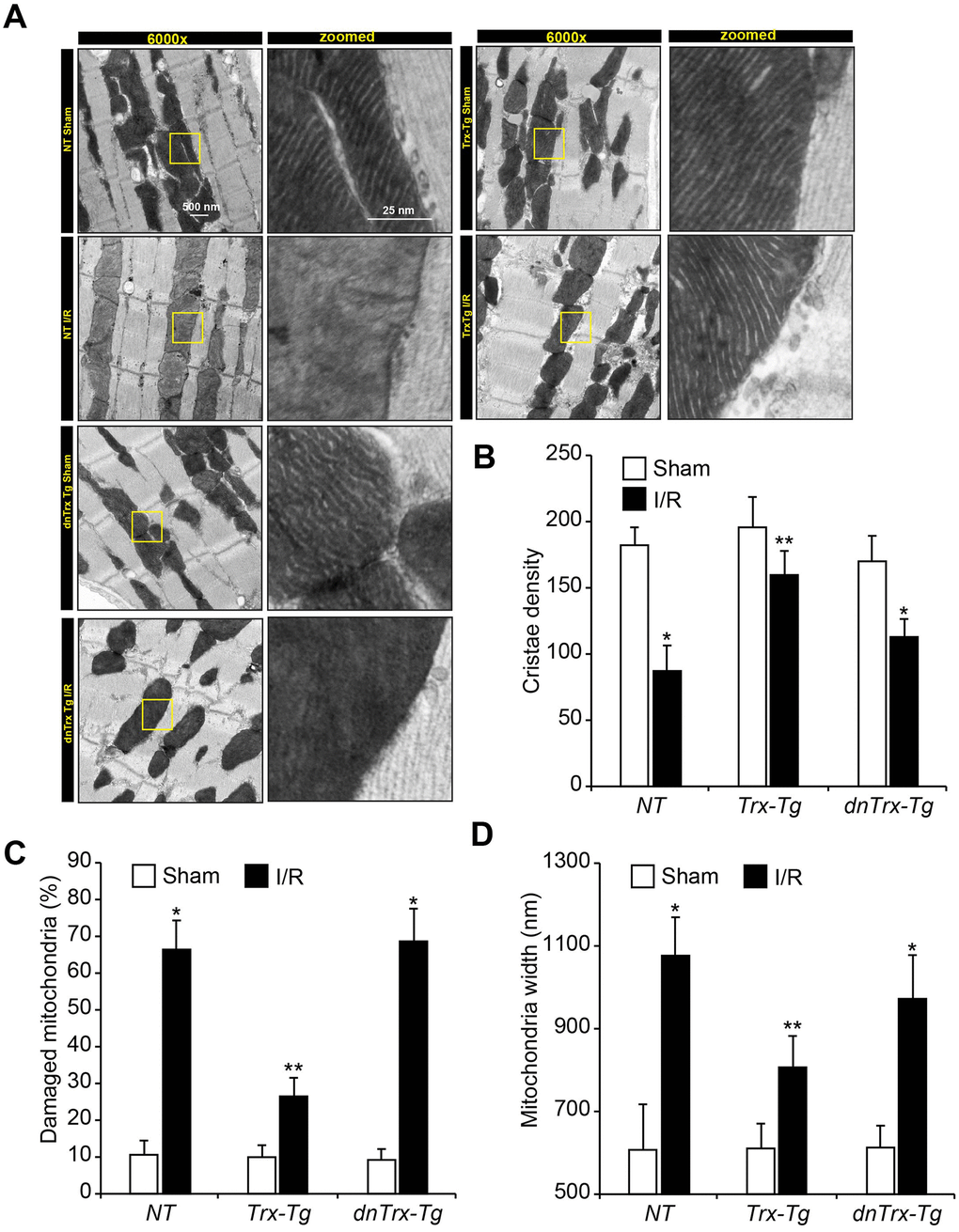
Figure 4. High level of Trx prevents I/R-induced mitochondrial cristae and DNA damage. (A) Ultrastructural analysis of sham or I/R hearts from NT, Trx-Tg and dnTrx-Tg. Representative transmission electron microscopic images showing cristae structure and density. Calculated cristae density (B), percent damaged mitochondria (loss of ≥50% cristae density) (C), and width of mitochondria (D) and plotted as bar graph. Values are means ± SD (n = 25). *, p < 0.01 versus Sham; **, p <0.01 versus NT or Trx-Tg IR.
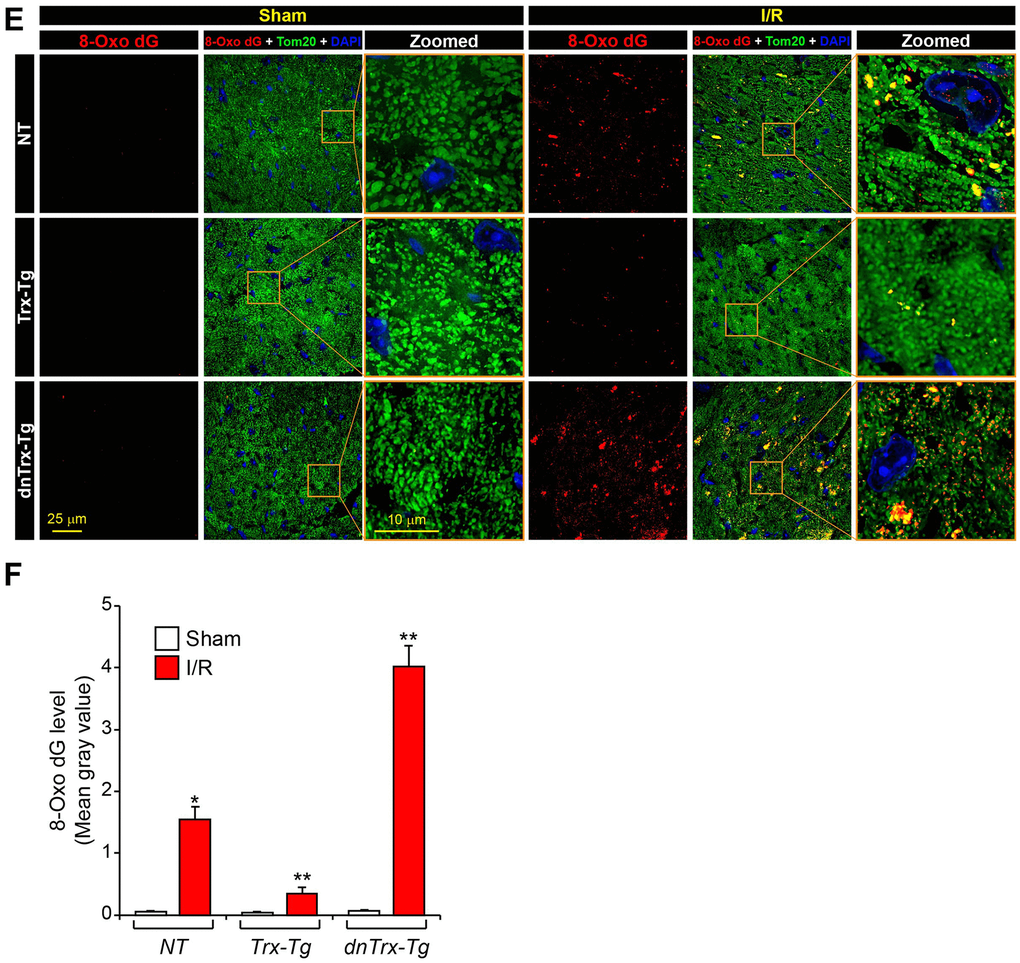
Figure 4. High level of Trx prevents I/R-induced mitochondrial cristae and DNA damage. (E) Immunofluorescence microscopic image shows accumulation of 8-oxo-dG in mitochondria of sham or I/R myocardium sections. (F) The levels of 8-oxo-dG was quantified and expressed as mean gray value. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R; † p <0.05 versus NT or Trx-Tg I/R. Statistical significance was determined with one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test.
Mitochondrial genome encodes the core proteins of ETC [26], and mitochondrial DNA damage is known to occur in I/R. [27] Therefore, we sought to determine whether high levels of Trx would ameliorate mitochondrial DNA damage in I/R. As shown in Figure 4E, 4F (top panels), increased levels of 8-Oxo-dG were observed in the heart sections in NT and dnTrx-Tg mice in I/R (Figure 4E, 4F, bottom panels), indicating significant mitochondrial DNA damage. Additionally, dnTrx-Tg mice showed about 2.5 fold higher level of 8-Oxo-dG level compared to NT mice, demonstrating a critical role of Trx in preservation of mitochondrial DNA in I/R stress. In contrast, Trx-Tg mice hearts had significantly lower levels of 8-Oxo-dG in I/R (Figure 4E, 4F, middle panels). These data demonstrate that Trx protects against mitochondrial DNA damage caused by I/R, which may protect mitochondrial ETC genes that, in turn, could preserve mitochondrial function in Trx-Tg mice in I/R.
Since mitochondrial cristae harbor the ETC, which carryout oxidative phosphorylation for supply of energy to the heart, we speculated a protective role of Trx in maintaining the oxidative phosphorylation due to preservation of mitochondrial structure in I/R, as aged mice show mitochondrial dysfunction in I/R [28]. We evaluated mitochondrial function by assessing ADP stimulated respiration and pyruvate-malate –dependent electron flow in mitochondria isolated from hearts of sham or I/R subjected mice. As shown in Figure 5A, 5B, succinate-driven state 2 respiration was significantly decreased in NT animals in I/R, but not in Trx-Tg mice (Figure 5E, 5F). Additionally, ADP-coupled state 3 respiration was decreased in I/R in NT or dnTrx-Tg mice, although there was no significant difference in Trx-Tg mice (Figure 5A, 5B, 5I, 5J and Figure 5E, 5F). Oligomycin-mediated state 4 respiration in NT or Trx-Tg mice was similar in sham animals, but I/R -subjected dnTrx-Tg mice had significant decrease in state 4 compared to either NT or Trx-Tg mice, demonstrating significant proton leak in dnTrx-Tg mice in I/R [29]. Collectively, these data demonstrate that electron flow might have been impaired that promoted disturbed proton gradient resulting in differential oxygen consumption. Therefore, we analyzed the flow of electrons via mitochondrial ETC complexes. Complex-I-mediated respiration utilizing pyruvate and malate as substrates was decreased in NT or dnTrx-Tg, but not in Trx-Tg mice (Figure 5C, 5D, 5G, 5H, 5K, 5L). However, complex-II-driven respiration by succinate was significantly lower in NT and dnTrx-Tg mice, but not in Trx-Tg mice. Further, ascorbate-TMPD driven complex IV OCR was decreased only in dnTrx-Tg mice in I/R, but not in NT or Trx-Tg mice, demonstrating significant impact of Trx in electron flow via mitochondrial complexes in I/R. Taken together, these data demonstrate significant protective effect of high levels of Trx on mitochondrial structure, function and energy metabolism.
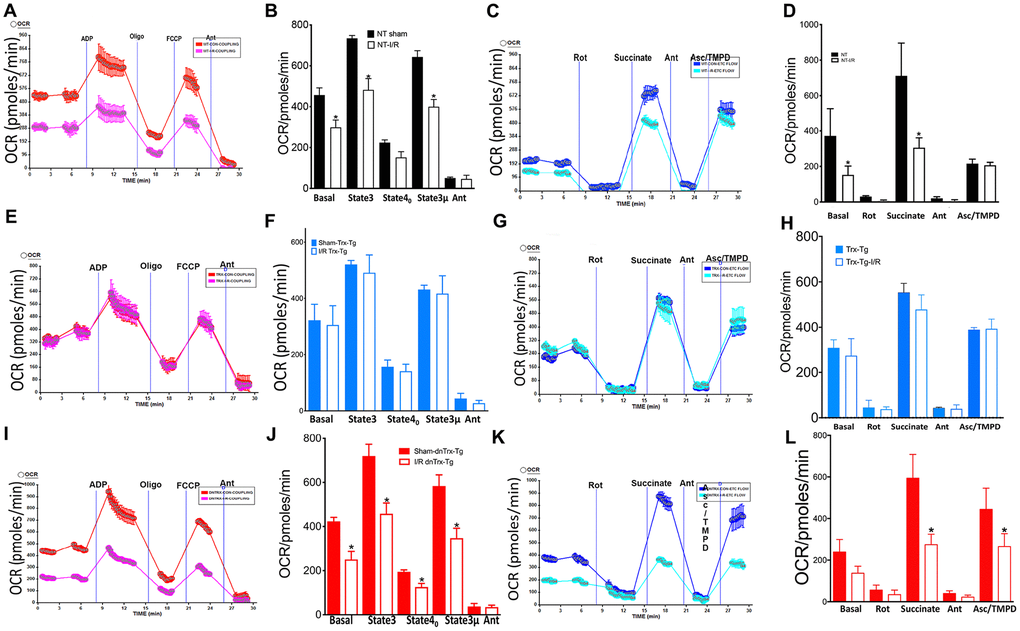
Figure 5. Trx protects against I/R-induced mitochondrial dysfunction. Effect of I/R on coupling and electron flow in mitochondria isolated from mice heart: Mitochondria were isolated from AAR region of sham or I/R subjected NT, Trx-Tg and dnTrx-Tg mice hearts as described in Materials and methods. (A) representative figure of XF24 coupling assay of sham or I/R mice mitochondria from (A) NT; (E) Trx-Tg; (I) dn-Trx-Tg mice, and OCR response to ADP, oligomycin, FCCP and antimycin A using point-to-point measurements. Graph of basal (state 2), state 3, and state 40 respirations in sham and I/R mice heart mitochondria from (B) NT; (F) Trx-Tg; (J) dn-TrxTg. A representative OCR measurement figure of mitochondria from (C) NT; (G) Trx-Tg; (K) dn-TrxTg mice with pyruvate and malate as substrates, and the effect of rotenone, succinate, antimycin A and Asc/TMPD. Sham and I/R mouse mitochondria from (D) NT; (H) Trx-Tg; (L) dn-TrxTg mice with basal, rotenone, succinate, antimycin A and Asc/TMPD mediated OCR (sham = 3, I/R n = 3). Statistical significance was determined with one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test. *P<0.05.
Trx prevents I/R -induced loss of mitochondrial proteins, by upregulating transcription of PGC1α
PGC1α is the master regulator of mitochondrial biogenesis and regulates several mitochondrial gene transcriptions as a coactivator of PPARγ. We analyzed the expression of mitochondrial proteins in mitochondrial extract from sham or I/R subjected mouse hearts. As shown Figure 6A, I/R did not change the expression of mitochondrial complexes, as analyzed by western blot using OXPHOS cocktail antibody (Abcam), which detects CI subunit NADH:ubiquinone oxidoreductase subunit8 (NDUFB8), complex II subunit 30kDa (CII-30kDa), CIII-Core protein 2, CIV subunit I and CV alpha subunit. We also analyzed aconitase (ACO2), mitofusin-1(MFN1), mitofusin-2(MFN2), Transcription factor A mitochondrial (TFAM), Hexokinase 1 (HK-1), superoxide dismutase-2 (Sod2) and cytochrome oxidase IV (COX IV) in mitochondrial extracts. As shown in Figure 6B, 6C, I/R resulted in significant loss of ACO2, MFN1 and MFN2 in NT or dnTrx-Tg mice in I/R mitochondrial extracts. However, high levels of Trx prevented I/R-induced loss of ACO2, MFN1 and MFN2 proteins (Figure 6B, 6C). I/R did not change the level of TFAM, HK-1, Sod2 and COX IV in NT, Trx-Tg, or dnTrx-Tg mice (Figure 6B, 6D).
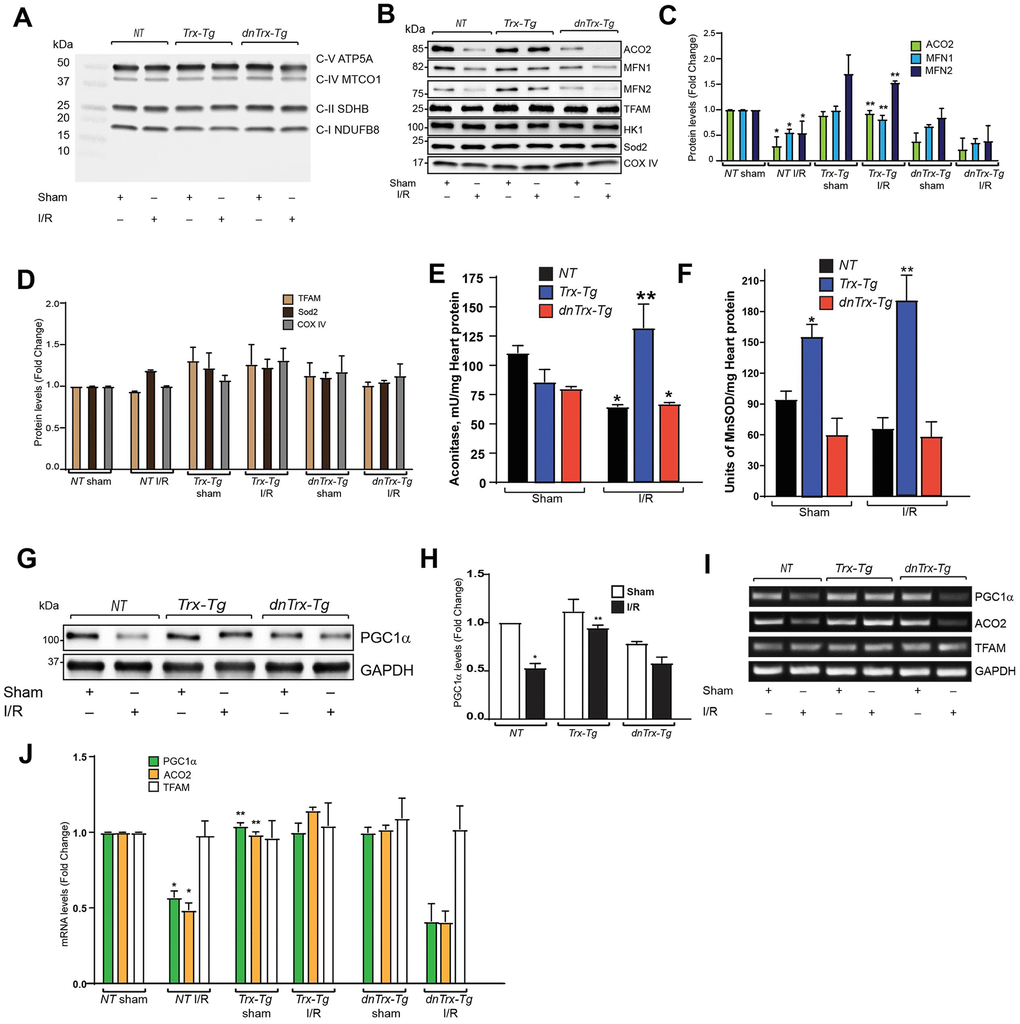
Figure 6. Trx prevents I/R-induced loss of mitochondrial proteins, by upregulating transcription of PGC1α. (A). Mitochondria was isolated from sham or I/R subjected NT, Trx-Tg and dnTrx-Tg mice. The mitochondrial extracts were analyzed for oxidative phosphorylation complex subunits by western blot using Abcam OXPHOS cocktail antibody. (B) Western blot analysis of ACO2, MFN1, MFN2, TFAM, Hexokinase 1 (HK-1), Sod2 and COX IV in sham or I/R mitochondrial extracts from NT, Trx-Tg and dnTrx-Tg mice. (C, D). Protein levels were quantified and expressed as fold change. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R. (E) Aconitase 2 activity and (F). MnSOD activity was determined in sham and I/R myocardium obtained from NT, Trx-Tg, or dnTrx-Tg mice as described in materials and methods. *p <0.05 versus respective sham; **p <0.05 versus NT or dnTrx-Tg I/R. n=3. (G) AAR region of sham or I/R myocardium from NT, Trx-Tg and dnTrx-Tg were lysed using M-PER lysis buffer and analyzed for PGC1α and GAPDH by western blotting. (H). Level of PGC1α was quantified and expressed as fold change. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R. (I). RT-PCR analysis of PGC1α, ACO2 and TFAM in sham and I/R myocardium (J). mRNA levels of PGC1a, ACO2 and TFAM were quantified and expressed as fold change. *p <0.05 versus NT sham; **p <0.05 versus NT or dnTrx-Tg I/R. Statistical significance was determined with the Student’s t test (C, D, H, and J) and one-way ANOVA followed by Tukey’s post-hoc multiple comparisons test (E, and F).
Since ACO2 protein expression was decreased in I/R, we sought to determine the activity of aconitase in mitochondrial extracts. As shown in Figure 6E, I/R resulted in loss of aconitase activity in NT and dnTrx-Tg mice, but not in Trx-Tg mice, which had significantly higher aconitase activity. We also evaluated Sod2 (MnSOD) activity in I/R, as it is an important enzyme of mitochondrial matrix that dismutates O2.- to H2O2, and thereby decreases mitochondrial oxidative stress. Although I/R did not alter the MnSOD activity in NT or dnTrx-Tg mice, Trx-Tg mice had significantly higher activity in sham or I/R subjected mice (Figure 6F). ACO2 activity was strongly correlated with its higher expression in Trx-Tg mice and decreased expression in NT or dnTrx-Tg mice in I/R. We determined whether the expression of PGC1α is modulated in I/R. As shown in Figure 6G, 6H, I/R caused significant loss of PGC1α expression in hearts of NT mice. In contrast, Trx-Tg mice were protected against I/R induced loss of PGC1α. Further, as shown in Figure 6I, 6J, I/R decreased mRNA expression of PGC1α and ACO2, but not TFAM in NT or dnTrx-Tg mice. In contrast, Trx-Tg mice had significantly higher level of PGC1α and ACO2. These data indicate that expression of PGC1α is essential for Trx-mediated protection of mitochondrial function.
Trx regulates expression of PGC1α via PI3K-AKT-CREB axis in cardiomyocytes
PGC1α expression is regulated by CREB, MEF-2 and ATF2 transcription factors [30]. Therefore, we determined whether expression of PGC1α and its upstream pathways are modulated by Trx redox state. As shown in Figure 7A, overexpression of Trx increased PGC1α levels in human coronary artery endothelial cells (HCAECs), but not in cells with redox-inactive Trx expression, indicating Trx-dependent redox regulation is critically important for PGC1α expression. Next, we determined the effect of Trx deficiency on PGC1α expression and CREB phosphorylation. Trx depletion inhibited hypoxia/reoxygenation (H/R) -induced expression of PGC1α and phosphorylation of CREB (Figure 7B). However, H/R did not downregulate the expression of PGC1α as observed in I/R (Figure 7B). Since major cell type in the heart is composed of endothelial cells, cardiomyocyte and fibroblast [31], we performed further studies with neonatal cardiomyocytes H9C2 to evaluated PGC1α regulation in cardiomyocytes. We have previously shown that MKK4 activation is regulated by Trx redox state [19]. Additionally, Trx activates MKK4 and PI3 kinase pathways and these signaling cascades are known to regulate PGC1α transcription via activation of CREB or ATF2 [19, 23]. As shown in Figure 7C, 7D, treatment of H9C2 with recombinant human Trx (rhTrx) activated MKK4, p38 and AKT. Pretreatment of rhTrx prevented H/R-induced loss of pAKT, PGC1α, MFN1 and MFN2, but potentiated the activation of MKK4, p38 and CREB (Figure 7C–7E). We also found that higher level of NRF1 transcription factor in rhTrx pretreated samples and there was no change in pATF2 level (Figure 7C, 7D). Since rescue of mitochondrial fusion proteins MFN1 and MFN2 was observed in rhTrx pretreated H9C2 cells exposed to H/R, we analyzed effect of Trx on proteins involved in mitochondrial fission, such as Drp1 or Fis1. As shown in Figure 7F, there was no change in the expression of Drp1 or Fis1 in H/R in presence or absence of Trx. Although our data demonstrate that both p38 and AKT are activated by rhTrx, we sought to delineate the specific pathway that activates Trx-mediated PGC1α expression in H/R. As shown in Figure 7G, inhibition of PI3 kinase blocked the Ad-Trx mediated expression of PGC1α and CREB activation, but not p38. Collectively, our data show that Trx regulates the expression of PGC1α via activation of PI3K-AKT-CREB axis. Further, elevated level of PGC1α in Trx treated cells or Trx-Tg mice upregulates the expression of ACO2, MFN1 and MFN2 by coactivating their transcription factors.
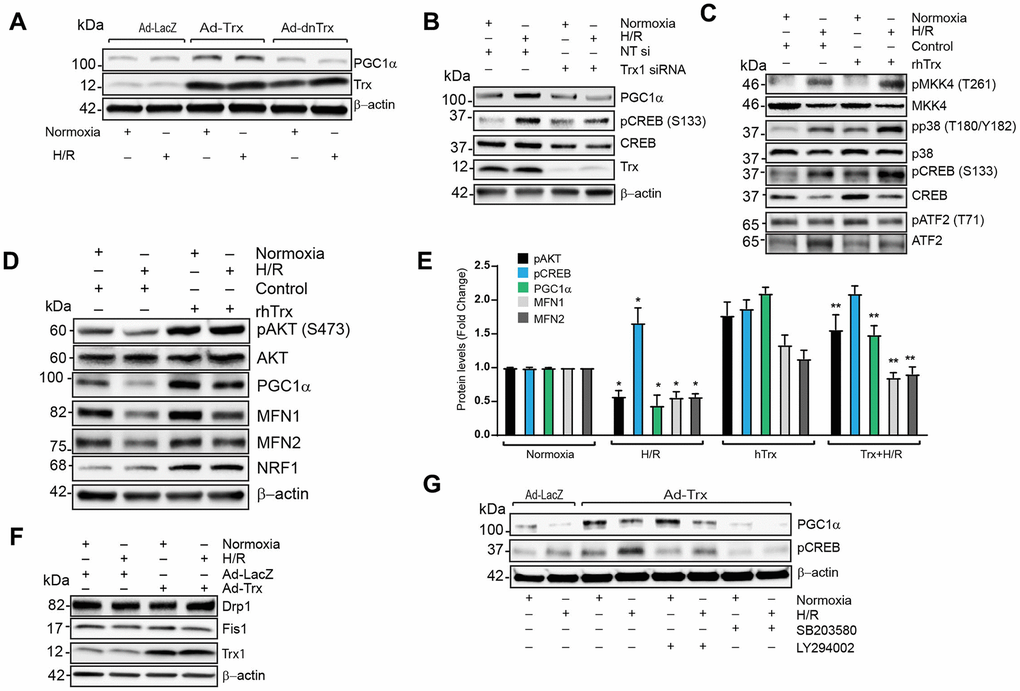
Figure 7. Trx regulates expression of PGC1α via PI3K-AKT-CREB axis in cardiomyocytes. (A). Western blot analysis of PGC1α, Trx and β-actin in lysate from Ad-LacZ, Ad-Trx and Ad-dnTrx infected and H/R (24/2h) exposed HCAECs. (B). Western blot analysis of PGC1α, pCREB (S133), CREB, Trx and b-actin in lysates from NT or Trx siRNA transfected and H/R (24/2h) exposed HCAECs. (C and D). Western blot analysis of pMKK4 (T261), MKK4, pp38 (T180/Y182), p38, pCREB (S133), CREB, pATF2 (T71), ATF2, pAKT (S473), AKT, PGC1α, MFN1, MFN2, NRF1 and β-actin in lysates from rhTrx pretreated and H/R (24/2h) exposed H9C2 cells. (E). Levels of protein in H9C2 cells were quantified and expressed as fold change. *p <0.05 versus Normoxia; **p <0.05 versus H/R. (F). Western blot analysis of Drp1, Fis1, Trx and β-actin in lysate from Ad-LacZ and Ad-Trx infected and H/R (24/2h) exposed H9C2 cells. (G). Western blot analysis of PGC1a, pCREB (S133) and β-actin in lysates from p38 inhibitor, SB203580 (2.5 μM) and PI3 kinase inhibitor, LY294002 (5.0 μM) pretreated and H/R (24/2h) exposed H9C2 cells. Statistical significance was determined with the Student’s t test.
Discussion
Aging is an independent risk factor for cardiovascular disorders including ischemic heart diseases [32]. A decline in cardiac mitochondrial function is a major contributor to age-related decrease in tolerance of the heart to stress such as I/R [6]. Continuous oxidative events over the life-span due to oxygen metabolism modifies mitochondrial proteins, impairing their critical role in energy production, and to overcome further oxidative insult such as I/R. Although ROS are unequivocally implicated in mitochondrial dysfunction in ischemic heart diseases such as I/R [4], antioxidant interventions have not only provided inconclusive results, but also failed to find their way into clinical translation. Accumulating evidence suggests that the mechanism of reperfusion injury such as myocardial infarction is complex and multi-factorial [33]. Oxidative protein modifications and mitochondrial dysfunction due to advancing age, compounded with I/R insult exert severe damage to the myocardium in aged individuals in I/R. Therefore, it is likely that one specific antioxidant might be ineffective to a multi-factorial damage to the aging heart in I/R injury.
The present study was undertaken to evaluate role of Trx in I/R injury in aged mice, as Trx is a unique multifunctional redox protein that regenerates oxidatively inactivated protein [17, 34], scavenges deleterious ROS such as hydroxyl radicals and singlet oxygen, induces mitochondrial superoxide dismutase-2 (SOD2) [18], and modulates various signal transduction pathways. We found that overexpression of Trx (2.5 to 3-fold) since the beginning of life prevents I/R injury in aged transgenic mice, as evidenced by reduced infarct size and decreased myocardial apoptosis. Trx improved left ventricular function by protecting against I/R -induced reduction in EF and FS. Additionally, high levels of Trx in Trx-Tg mice rescued structural and functional impairment of mitochondria and improved mitochondrial energy metabolism in I/R. Using Trx-deficient mice (dnTrx-Tg), we found that the decreased levels of active Trx (about 4-fold decrease in activity) exacerbate I/R-induced infarct size and myocardial apoptosis. Further, Trx prevented I/R-induced loss of PGC1α mRNA and protein expression, resulting in protection of I/R-mediated decreased expression of ACO2, MFN1, and MFN2. Further, Trx-mediated activation of AKT-CREB-PGC1α signaling cascade is essential for the regulation of mitochondrial gene expression and function in I/R.
Three-fold higher expression of Trx in mice prevented oxidation of Trx in I/R, and showed increased TrxR1 activity, preserving the redox state of myocardium during I/R insult in contrast to mice with decreased levels of active Trx. This is important for further protective role of Trx as Trx itself undergoes oxidation and other modifications. Oxidative modifications of mitochondrial proteins, such as carbonylation, S-glutathionylation, and accumulation of protein disulfides and mixed disulfides in the aged myocardium alter the structure and function of mitochondria during the aging process. [35] These modifications lead to decline in mitochondrial respiration, mitochondrial content, and the accumulation of defective mitochondria resulting in ROS production, oxidative injury, and cell death [6]. Aging heart with these incapacitated mitochondria acutely fails to overcome or adapt to I/R insult resulting in life-threatening myocardial infarction [9]. We have previously shown that mitochondrial succinate-dependent energy coupling with addition of ADP significantly declines in aged mice [6]. Additionally, the electron transfer due to oxidation of pyruvate/malate via complex I to IV significantly decreases in aged mice [6]. Further, loss of aconitase expression and activity in I/R in NT or dnTrx-Tg mice, but not in Trx-Tg mice potentially decrease NADH production and loss of electron flow. A previous study has shown decreased aconitase activity without decrease expression in rat I/R model, where the entire heart tissue was evaluated after I/R in ex vivo Langendorff isolated heart model with retrograde perfusion. [36] In contrast, in our study, we evaluated mitochondrial function in vivo and found that decreased aconitase expression in the infarcted area in I/R. Additionally, studies have demonstrated that α-ketoglutarate dehydrogenase (αKGDH) and aconitase of TCA cycle are inactivated in I/R due to oxidation of critical sulfhydryl and inactivation of [4Fe-4S]2+ cluster center in aconitase [37]. Since Trx reduces oxidized proteins, and we did not observe decreased aconitase activity or decreased electron flow in Trx-Tg mice, it is likely that high levels of Trx might have reversed aconitase and αKGDH to their native functional state due to its disulfide reducing properties. We found that mitochondrial coupling and electron transfer via complex I to complex IV remains functional in the mitochondria isolated from Trx-Tg mice in I/R, but not from NT or dnTrx-Tg mice, which is correlated with protection of aconitase expression and activity in Trx-Tg mice, indicating a redox-related mechanism. In this regard, aconitase activity was restored in aged Trx-Tg mice in I/R, but not in dnTrx-Tg mice. Further, the activity of Sod2 was increased in Trx-Tg mice in I/R in contrast to dnTrx-Tg mice, indicating loss of antioxidative function due to lack of redox-active Trx.
Enhancing mitochondrial fusion or preventing the mitochondrial fission has been shown to protect against I/R injury [10]. Conditional knock out of Mfn1 and Mfn2 in adult hearts induced mitochondrial fragmentation, mitochondrial respiratory dysfunction, which lead to dilated cardiomyopathy [38]. Additionally, previous studies have shown that loss of mitofusin during I/R constitutes a mechanism of I/R injury. [39] In the present study, we demonstrated that Trx prevented the loss of MFN1 and 2, which could be another mechanism related to Trx-mediated protection of mitochondrial dysfunction in I/R. Collectively our data show that 2.5 to 3-fold increase in Trx prevents loss of expression of MFNs and aconitase and thus preserved the mitochondrial structure and function during I/R.
PGC1α regulates the expression of several mitochondrial genes via its binding to transcription factors such as ERRs, PPARs, and NRFs [13]. The PGC1α KO mouse shows decreased expression of mitochondrial oxidative phosphorylation genes, mitochondrial enzymatic activities and reduced levels of ATP [40]. In the present study, we found decreased PGC1α mRNA and protein expression in I/R, which was restored by Trx overexpression. Consistent with this finding an earlier study has shown upregulation of PGC1α by Trx in mouse heart [41]. Although unclear, we speculate that this finding could be due to the cardiac-specific expression of PGC1 α and the use of the ex vivo Langendorff global ischemia model. In our in vivo and cell culture studies we found that PGC1α expression was inhibited by I/R or H/R, respectively. Since PGC1α transcription is regulated by CREB [30], ATF2 [42], and MEF2 [43] transcription factors, we found that Trx upregulates PGC1α expression by activation of the AKT-CREB signaling. In a previous study we have demonstrated the activation of AKT by Trx [23].
In conclusion, our data established that high levels of Trx preserved Trx redox state and mitochondrial structure, resulting in mitochondrial integrity in I/R. Additionally, Trx rescued aconitase and Sod2 activity via its disulfide reductase properties that allowed uninterrupted mitochondrial energy production during I/R via enhanced coupling and flow of electrons, and substrate oxidation via functional mitochondria. High levels of Trx also promoted mitochondrial biogenesis by promoting the expression of PGC1α via PI3K-AKT-CREB pathway during I/R. Upregulation of P13K-AKT-CREB- PGC1α axis prevents the loss of MFN1, MFN2, ACO2 and also maintained the activity of aconitase resulting in improved mitochondrial function and decreased apoptosis.
Materials and Methods
Antibodies and chemicals
Antibodies and chemicals were obtained from following vendors. Abcam (Cambridge, MA): Total OXPHOS Rodent WB Antibody Cocktail (ab110413), anti-aconitase 2 (110320), anti-Hexokinase 1 (ab150423) and anti-TOMM20 (ab205486); BD Bioscience: Anti-Drp1 (611738) and anti-Cytochrome C (556433); Cell Signaling Technologies (Danvers, MA): Anti-Trx (2298), anti-Cleaved caspase-3 (9661), anti-Bax (2772), anti- Phospho-CREB (Ser133) (9191), anti-Phospho-Akt (Ser473) (4060), anti-AKT (9272), anti-Phospho-SEK1/MKK4 (Thr261) (9151), anti-SEK1/MKK4 (9152), anti-Phospho-p38 MAPK (Thr180/Tyr182) (4511), anti p38 MAPK Antibody (9212) and anti-Phospho-ATF-2 (Thr71) Antibody (9221); Santa Cruz Biotechnology (Dallas, TX): Anti-Actin (sc-1616), anti-Mfn1 Antibody (sc-50330), anti-COX4 (sc-58348), anti-NRF-1 Antibody (sc-33771), anti-ATF-2 Antibody (sc-187) and anti-Fis1 Antibody (sc-98900); Novus Biologicals (Centennial, CO): anti- PGC1 alpha Antibody (NBP1-04676); Sigma (St. Louis, MO): Anti-MFN2 antibody (WH0009927M3), Anti-MnSOD Antibody (06-984), Anti-8-Oxoguanine Antibody (MAB3560), anti-TFAM, anti-GAPDH antibody-HRP conjugate, anti β-actin-HRP conjugate, and recombinant human Trx; Thermo Scientific (Waltham, MA): Alexa Fluor 488, 568, 647-conjugated secondary antibodies, secondary anti-rabbit, anti-mouse IgG-HRP antibodies, isolectin IB4-Alexa Fluor 568 and M-PER Mammalian Protein Extraction Reagent; All other chemicals were purchased from Sigma unless otherwise stated.
Animals and cells
Wild-type C57BL6 strain (WT) were purchased from Charles River Laboratory. Transgenic mice with overexpression of human Trx (Trx-Tg) or dominant-negative Trx (dnTrx-Tg) were bred and maintained in the animal facility of Texas Tech University Health Sciences Center and have been described previously [25]. Both males and females were used in this study. All mice strains used in this study are from a C57BL/6 background and are 20-26 months of age, equivalent to human age of 70-75 years [44]. All animal procedures were approved by the Institutional Animal Care and Use Committee (IACUC) of the Texas Tech University Health Sciences Center and were consistent with the Guide for the Care and Use of Laboratory Animals published by the National Institute of Health. HCAEC were purchased from Clonetics and propagated in endothelial basal medium supplemented with additives (Bullet kit, Clonetics). H9c2 cells were purchased from ATCC and propagated in DMEM with 10% FBS.
Cell culture and hypoxia/ reoxygenation (H/R)
HCAECs and H9C2 cells in complete medium were flushed with a 95% N2, 5% CO2 gas mixture while in a Billups-Rothenberg modular chamber to create a hypoxic environment. The oxygen level was kept below 1% by measuring with an oxygen electrode. Chambers were kept inside the incubator at 37 °C for indicated periods of time and followed by 2 h of reoxygenation in normoxic condition.
Myocardial ischemia and reperfusion
NT, Trx-Tg and dnTrx-Tg littermates were anesthetized with ketamine (100 mg/kg) and xylazine (10 mg/kg), by injecting via intra peritoneal route (i.p.). After an equilibration period of 10 min, the left thoracotomy was performed in the fourth intercostal space, and the pericardium was opened to expose the heart. An 8-0 silk suture was passed around the LAD at a point two-thirds of the way between its origin near the pulmonary conus and the cardiac apex. Coronary artery occlusion was achieved by ligating the left descending coronary artery using a slipknot. Following 60 minutes of ischemia, the slipknot was released, and the myocardium was reperfused for 30 minutes. Sham mice underwent the same procedure without the slipknot tied. Mice were sacrificed after 60 minutes of ischemia followed by 30 minutes of reperfusion. To collect heart samples, mice were euthanized by injecting ketamine (100 mg/kg) and xylazine (10 mg/kg) via i.p route.
Adenovirus production
AdenoX system was obtained from Stratagene Corp. (La Jolla, CA), and LacZ or Trx cDNA was cloned into pAdenoX vector as described previously [45]. Recombinant virus was allowed to infect HEK293 cells for generation of viral particles.
Determination of infarct size
Myocardial infarct size was determined as described previously [46]. Briefly, after reperfusion, animals were sacrificed, and the aortae were cannulated and perfused with saline to remove blood. 0.25 ml of 1.5% Evans blue was perfused after religating the coronary artery to demarcate remote myocardium (blue) and AAR. 1.0-mm heart sections were made using mouse coronal matrix and stained with 1.0% triphenyltetrazolium chloride (TTC) for 15 min at 37 °C. After TTC staining, paraformaldehyde-fixed heart sections were photographed using Nikon D5200 camera using Nikon AF-S DX NIKKOR 18-55 mm lens at f/6.3, 1/160s, ISO200. TTC stained and unstained area (infarct) at AAR was quantified using Adobe Photoshop.
Myocardial echocardiography
Transthoracic echocardiography was performed on anesthetized mice using a Visual Sonics Vevo3100 (Toronto, ON, Canada) Imaging System with a 30-MHz high-frequency transducer (MX400). After sham or I/R surgery, mouse under Ketamine and Xylazine anesthesia was laid supine on a heated platform, and M-mode images were recorded at the level of the papillary muscle. The left ventricular ejection fraction (EF%) and fractional shortening (FS %) were calculated using Vevo LAB software.
Electron microscopy
Sham and I/R subjected hearts were immediately fixed by a retrograde perfusion with 2% glutaraldehyde in PBS. Then, 1.0 mm thick sections were prepared from AAR region and stored in 4% glutaraldehyde in PBS. Ultrathin sectioning was completed in Electron Microscopy Core Facility, UT Southwestern Medical Center, Dallas, TX. Ultrastructure images from copper grid mounted ultrathin section of samples were obtained using Hitachi High-Technologies H-7650 Transmission electron microscope in College of Arts and Sciences Microscopy, Texas Tech University, Lubbock, TX. Mitochondrial cristae density was calculated from the inverse of calibrated mean gray value from the electron-dense area inside the inner-mitochondrial membrane of intrafibrillar mitochondria. Total and damaged mitochondria numbers were counted from 6,000x magnification TEM images, and percent damaged mitochondria was calculated. Mitochondria with loss of ≥50% cristae density were considered damaged mitochondria. Mitochondria width was calculated from 10,000x TEM images.
RNA interference
Small interfering RNAs were obtained for nontargeting siRNA control, and hTrx from Dharmacon Inc. (Arvada, CO). 100 nM of siRNA was transfected using lipofectamine RNAiMAX reagent obtained from Thermo Scientific (Waltham, MA). Inhibition of gene expression by siRNA was determined after 36-48 h by Western blotting.
RT-PCR
Total RNA was isolated from RNAlater preserved AAR region of mouse hearts using TRIzol (Cat. No. 15596-018, Ambion), and cDNA was generated by reverse transcription reaction using high-capacity cDNA Reverse Transcription Kit (Cat. No. 4374966, Applied Biosystems). The cDNA was then used as a template for PCR amplification using the following primers:
Western blotting
Protein extracts were prepared from appropriately treated H9C2, HCAEC cells or sham or infarcted left ventricle using M-PER mammalian protein extraction reagent from Thermo Scientific (Waltham, MA) with protease and phosphatase inhibitors. The homogenates were centrifuged at 10, 000 rpm for 10 min at 4°C. Protein concentrations were determined with the BCA protein assay kit (Pierce Chemical, Rockford, IL). For analysis of Cyt-C, cytosolic and mitochondrial extracts were prepared using Abcam Mitochondria Isolation Kit for Tissue (ab110168). Protein extracts were analyzed by Western blotting using their specific antibodies.
| Gene | Forward Primer | Reverse Primer | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mACO2 | GGTGGCTGTACCATCAACCA | TTCACACCGATCACCTTGGG | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mPGC1α | TTGGTGACCATGACTACTGT | AAGTCTCTCTCAGGTAGCAC | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mTFAM | GTCACGAAGCTCAGTAGGCA | CTCCACAGGGCTGCAATTTT | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| mGAPDH | CCAGAACATCATCCCTGCAT | CATCGAAGGTGGAAGAGTGG | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The amplified PCR products were separated on 2% agarose gels and stained with ethidium bromide. Gel images were captured using LI-COR odyssey Fc Imaging System. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Trx redox state assay
Carboxymethylation of mice heart tissue was performed as described in our previous publications [15, 25]. Briefly, AAR region of heart tissue (10–20 mg) was homogenized in 0.5 ml carboxymethylation buffer (0.1 M Tris·HCl pH 8.8, 6 M guanidine hydrochloride, and 10 mg/ml iodoacetic acid), and after the addition of 5 μl of 10% Triton X-100 the samples were incubated for 1 h at 37°C in the dark. Samples were centrifuged in a tabletop refrigerated centrifuge (Eppendorf) for 10 min at 2,500 rpm, and the supernatants (0.5 ml) were transferred to desalting columns to remove guanidine hydrochloride. Protein content was determined by the Bradford method (Bio-Rad, Hercules, CA). Twenty micrograms of carboxymethylated heart tissue homogenate were fractionated on a 15% native polyacrylamide gel (Bio-Rad). Transferred to nitrocellulose membrane and probed with anti-Trx antibody.
Immunofluorescence microscopy
Sham or I/R heart tissue sections were deparaffinized, hydrated, permeabilized, blocked, and immunostained with anti-8-Oxo-dG and anti-Tom 20 antibodies followed by Alexa Fluor 488- and Alexa Fluor 568-conjugated donkey anti-rabbit and anti-mouse secondary antibodies. Nuclei were counterstained with DAPI. Fluorescence images were obtained via 100x/1.4 NA objective using Zeiss Axio Imager Z2 upright fluorescent microscope. The fluorescence intensity was quantitated using Adobe Photoshop.
Mitochondria isolation for XF24 assay
AAR region of sham or I/R hearts were surgically removed. Mitochondria from hearts of mice were isolated as described earlier [7]. 80 mg of AAR region of heart was minced and homogenized at 4 °C using Kimble Chase 2mL tissue grinder tube with sequential use of pestle A and pestle B in mitochondrial isolation buffer (70 mM sucrose, 210 mM mannitol, 5 mM HEPES, pH 7.2, 1 mM EGTA and 0.5% fatty acid free BSA). The homogenate was centrifuged at 5,000 ×g for 10 min. The supernatant was again centrifuged at 1600 g for 5 min. The supernatant was centrifuged at 12500g for 10 min at 4°C in Avanti J-E centrifuge using JA20 rotor. The translucent white pellet was resuspended gently in buffer A and centrifuged again in Avanti J-E centrifuge at 25000g for 5 min at 4°C. The mitochondrial pellet was suspended in mitochondrial isolation buffer without BSA and protein was estimated with Bradford assay (Biorad, Rockford, IL). Mitochondria were suspended at 1.5 μg/50 μl in 1× mitochondrial assay buffer (MAS; 70 mM sucrose, 220 mM mannitol, 10 mM KH2PO4, 5 mM MgCl2, 2 mM HEPES, 1 mM EGTA, and 0.2% fatty acid free BSA; pH 7.2 at room temperature) and plated into each well of the v7 assay plate of XF24 analyzer.
XF24 instrument setup and analysis
Analysis of mitochondrial function was performed in XF24 flux analyzer (Seahorse, Bellerica, MA) as previously published. [7] Briefly, XF24 instrument was equilibrated at 37°C overnight. 1.5μg of mouse heart mitochondria was plated in each well of the XF24 v7 plate in a volume of 50 μl containing 1× MAS with 10 mM succinate and 2 μM rotenone as substrate for coupling assay; and 10 mM pyruvate, 2 mM malate and 4 μM FCCP was added to 1× MAS for the electron flow experiment as described in our previous publication [7].
Thioredoxin and thioredoxin reductase assays
Trx and Trx reductase activity assay were performed in sham or myocardium as described in our previous publication [47]. All assays were performed in Beckman DU800 spectrometer with temperature control and using quartz cuvettes.
Aconitase and SOD2 assays
Aconitase activity was measured in isolated mitochondria as described before [48] and SOD2 activity as described in our previous publication [7].
Quantification of apoptosis in heart by EPR
To quantify total apoptosis in the infarcted tissue of mouse heart, we modified an assay method originally developed by Fabisiak et al. [49], using annexin-V magnetic microbeads kit from Miltenyi Biotec GmbH, Germany (Cat. No. 130-090-201). After sham or IR surgery, the heart was quickly isolated from mice, cannulated via aortic arch and perfused with ice cold saline followed by 1x annexin-V binding buffer supplied by the manufacturer. Following complete removal of circulating blood, the heart was perfused with 250 μL of annexin-V microbead suspension and incubated at 2-4°C for 20 minutes. At the end of the incubation period, the heart was perfused with ice cold 1x annexin-V binding buffer and the entire infarcted tissue (area lower to the occlusion site in the LAD) was dissected out. Total annexin-V bound to infarcted tissue was quantified by measuring conjugated iron spins using Bruker EMX Micro spectrometer at room temperature. EPR spectra were acquired under following scan conditions: microwave frequency, 9.83 GHz; power, 30 mW; attenuation 8 dB; modulation frequency, 100 kHz; modulation amplitude, 4.00 G; sweep time, 60 s; time constant, 20.48 s; receiver gain, 20 dB; magnetic field, 2110-4110 G. Absolute spin counts from spectra were calculated using Quantitative EPR module of Bruker Xenon Micro 1.3 software.
Statistical analysis
The experiments were performed in triplicate and repeated for a minimum of 2 times. All cell culture studies were performed in triplicate and repeated at least twice. Data were statistically analyzed by analysis of variance for multiple means with Tukey's post hoc analysis. Student's t-test was used to compare two means. Prism software (Version 8.0) was used for all statistical analyses.
Author Contributions
KCD conceptualized and designed the research, performed Trx and TrxR activity assays, mitochondrial function analysis by flux analyzer, analyzed data, wrote and edited the manuscript; JS designed and performed cardiac function analysis by echocardiography, designed and performed all western analysis including PGC1α, AKT phosphorylation and electron microscopic analysis of mitochondrial structure, analyzed data and wrote manuscript; VKS, performed EPR apoptosis experiments, prepared the grid for EM analysis, immunofluorescence of 8-Oxo-dG levels and quantifications and analyzed mitochondrial structural parameters and quantification.
Conflicts of Interest
The authors have no conflicts to declare.
Funding
The work is supported by National Heart Lung and Blood Institute of National Institutes of Health grant number HL107885, HL109397 and HL 132953 to K.Das.
References
- 1. Susic D, Frohlich ED. The aging hypertensive heart: a brief update. Nat Clin Pract Cardiovasc Med. 2008; 5:104–10. https://doi.org/10.1038/ncpcardio1091 [PubMed]
- 2. Liu P, Xu B, Cavalieri TA, Hock CE. Attenuation of antioxidative capacity enhances reperfusion injury in aged rat myocardium after MI/R. Am J Physiol Heart Circ Physiol. 2004; 287:H2719–27. https://doi.org/10.1152/ajpheart.00317.2004 [PubMed]
- 3. Santos CX, Raza S, Shah AM. Redox signaling in the cardiomyocyte: from physiology to failure. Int J Biochem Cell Biol. 2016; 74:145–51. https://doi.org/10.1016/j.biocel.2016.03.002 [PubMed]
- 4. Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res. 2014; 114:524–37. https://doi.org/10.1161/CIRCRESAHA.114.300559 [PubMed]
- 5. Moris D, Spartalis M, Spartalis E, Karachaliou GS, Karaolanis GI, Tsourouflis G, Tsilimigras DI, Tzatzaki E, Theocharis S. The role of reactive oxygen species in the pathophysiology of cardiovascular diseases and the clinical significance of myocardial redox. Ann Transl Med. 2017; 5:326. https://doi.org/10.21037/atm.2017.06.27 [PubMed]
- 6. Lesnefsky EJ, Chen Q, Hoppel CL. Mitochondrial metabolism in aging heart. Circ Res. 2016; 118:1593–611. https://doi.org/10.1161/CIRCRESAHA.116.307505 [PubMed]
- 7. Das KC, Muniyappa H. Age-dependent mitochondrial energy dynamics in the mice heart: role of superoxide dismutase-2. Exp Gerontol. 2013; 48:947–59. https://doi.org/10.1016/j.exger.2013.06.002 [PubMed]
- 8. Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev. 2014; 94:909–50. https://doi.org/10.1152/physrev.00026.2013 [PubMed]
- 9. Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL. Mitochondrial dysfunction and myocardial ischemia-reperfusion: implications for novel therapies. Annu Rev Pharmacol Toxicol. 2017; 57:535–65. https://doi.org/10.1146/annurev-pharmtox-010715-103335 [PubMed]
- 10. Maneechote C, Palee S, Kerdphoo S, Jaiwongkam T, Chattipakorn SC, Chattipakorn N. Balancing mitochondrial dynamics via increasing mitochondrial fusion attenuates infarct size and left ventricular dysfunction in rats with cardiac ischemia/reperfusion injury. Clin Sci (Lond). 2019; 133:497–513. https://doi.org/10.1042/CS20190014 [PubMed]
- 11. Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation. 2010; 121:2012–22. https://doi.org/10.1161/CIRCULATIONAHA.109.906610 [PubMed]
- 12. Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. 2017; 360:12–18. https://doi.org/10.1016/j.yexcr.2017.03.052 [PubMed]
- 13. Di W, Lv J, Jiang S, Lu C, Yang Z, Ma Z, Hu W, Yang Y, Xu B. PGC-1: The Energetic Regulator in Cardiac Metabolism. Curr Issues Mol Biol. 2018; 28:29–46. https://doi.org/10.21775/cimb.028.029 [PubMed]
- 14. Valle I, Alvarez-Barrientos A, Arza E, Lamas S, Monsalve M. PGC-1alpha regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005; 66:562–73. https://doi.org/10.1016/j.cardiores.2005.01.026 [PubMed]
- 15. Muniyappa H, Song S, Mathews CK, Das KC. Reactive oxygen species-independent oxidation of thioredoxin in hypoxia: inactivation of ribonucleotide reductase and redox-mediated checkpoint control. J Biol Chem. 2009; 284:17069–81. https://doi.org/10.1074/jbc.M109.008557 [PubMed]
- 16. Das KC, Pahl PM, Guo XL, White CW. Induction of peroxiredoxin gene expression by oxygen in lungs of newborn primates. Am J Respir Cell Mol Biol. 2001; 25:226–32. https://doi.org/10.1165/ajrcmb.25.2.4314 [PubMed]
- 17. Holmgren A, Björnstedt M. Thioredoxin and thioredoxin reductase. Methods Enzymol. 1995; 252:199–208. https://doi.org/10.1016/0076-6879(95)52023-6 [PubMed]
- 18. Das KC, Lewis-Molock Y, White CW. Elevation of manganese superoxide dismutase gene expression by thioredoxin. Am J Respir Cell Mol Biol. 1997; 17:713–26. https://doi.org/10.1165/ajrcmb.17.6.2809 [PubMed]
- 19. Kundumani-Sridharan V, Subramani J, Das KC. Thioredoxin activates MKK4-NFκB pathway in a redox-dependent manner to control manganese superoxide dismutase gene expression in endothelial cells. J Biol Chem. 2015; 290:17505–19. https://doi.org/10.1074/jbc.M115.660365 [PubMed]
- 20. Huang Q, Zhou HJ, Zhang H, Huang Y, Hinojosa-Kirschenbaum F, Fan P, Yao L, Belardinelli L, Tellides G, Giordano FJ, Budas GR, Min W. Thioredoxin-2 inhibits mitochondrial reactive oxygen species generation and apoptosis stress kinase-1 activity to maintain cardiac function. Circulation. 2015; 131:1082–97. https://doi.org/10.1161/CIRCULATIONAHA.114.012725 [PubMed]
- 21. Tao L, Gao E, Hu A, Coletti C, Wang Y, Christopher TA, Lopez BL, Koch W, Ma XL. Thioredoxin reduces post-ischemic myocardial apoptosis by reducing oxidative/nitrative stress. Br J Pharmacol. 2006; 149:311–18. https://doi.org/10.1038/sj.bjp.0706853 [PubMed]
- 22. Subramani J, Kundumani-Sridharan V, Hilgers RH, Owens C, Das KC. Thioredoxin uses a GSH-independent route to deglutathionylate endothelial nitric-oxide synthase and protect against myocardial infarction. J Biol Chem. 2016; 291:23374–89. https://doi.org/10.1074/jbc.M116.745034 [PubMed]
- 23. Hilgers RH, Kundumani-Sridharan V, Subramani J, Chen LC, Cuello LG, Rusch NJ, Das KC. Thioredoxin reverses age-related hypertension by chronically improving vascular redox and restoring eNOS function. Sci Transl Med. 2017; 9:eaaf6094. https://doi.org/10.1126/scitranslmed.aaf6094 [PubMed]
- 24. Tao L, Gao E, Bryan NS, Qu Y, Liu HR, Hu A, Christopher TA, Lopez BL, Yodoi J, Koch WJ, Feelisch M, Ma XL. Cardioprotective effects of thioredoxin in myocardial ischemia and reperfusion: role of s-nitrosation [corrected]. Proc Natl Acad Sci USA. 2004; 101:11471–76. https://doi.org/10.1073/pnas.0402941101 [PubMed]
- 25. Das KC. Thioredoxin-deficient mice, a novel phenotype sensitive to ambient air and hypersensitive to hyperoxia-induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2015; 308:L429–42. https://doi.org/10.1152/ajplung.00285.2014 [PubMed]
- 26. Bibb MJ, Van Etten RA, Wright CT, Walberg MW, Clayton DA. Sequence and gene organization of mouse mitochondrial DNA. Cell. 1981; 26:167–80. https://doi.org/10.1016/0092-8674(81)90300-7 [PubMed]
- 27. Yue R, Xia X, Jiang J, Yang D, Han Y, Chen X, Cai Y, Li L, Wang WE, Zeng C. Mitochondrial DNA oxidative damage contributes to cardiomyocyte ischemia/reperfusion-injury in rats: cardioprotective role of lycopene. J Cell Physiol. 2015; 230:2128–41. https://doi.org/10.1002/jcp.24941 [PubMed]
- 28. Lesnefsky EJ, Hoppel CL. Ischemia-reperfusion injury in the aged heart: role of mitochondria. Arch Biochem Biophys. 2003; 420:287–97. https://doi.org/10.1016/j.abb.2003.09.046 [PubMed]
- 29. Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda). 2011; 26:192–205. https://doi.org/10.1152/physiol.00046.2010 [PubMed]
- 30. Herzig S, Long F, Jhala US, Hedrick S, Quinn R, Bauer A, Rudolph D, Schutz G, Yoon C, Puigserver P, Spiegelman B, Montminy M. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001; 413:179–83. https://doi.org/10.1038/35093131 [PubMed]
- 31. Zhou P, Pu WT. Recounting cardiac cellular composition. Circ Res. 2016; 118:368–70. https://doi.org/10.1161/CIRCRESAHA.116.308139 [PubMed]
- 32. Steenman M, Lande G. Cardiac aging and heart disease in humans. Biophys Rev. 2017; 9:131–37. https://doi.org/10.1007/s12551-017-0255-9 [PubMed]
- 33. Eltzschig HK, Eckle T. Ischemia and reperfusion—from mechanism to translation. Nat Med. 2011; 17:1391–401. https://doi.org/10.1038/nm.2507 [PubMed]
- 34. Holmgren A, Johansson C, Berndt C, Lönn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005; 33:1375–77. https://doi.org/10.1042/BST20051375 [PubMed]
- 35. Mailloux RJ, Willmore WG. S-glutathionylation reactions in mitochondrial function and disease. Front Cell Dev Biol. 2014; 2:68. https://doi.org/10.3389/fcell.2014.00068 [PubMed]
- 36. Sadek HA, Humphries KM, Szweda PA, Szweda LI. Selective inactivation of redox-sensitive mitochondrial enzymes during cardiac reperfusion. Arch Biochem Biophys. 2002; 406:222–28. https://doi.org/10.1016/s0003-9861(02)00446-0 [PubMed]
- 37. Bulteau AL, Lundberg KC, Ikeda-Saito M, Isaya G, Szweda LI. Reversible redox-dependent modulation of mitochondrial aconitase and proteolytic activity during in vivo cardiac ischemia/reperfusion. Proc Natl Acad Sci USA. 2005; 102:5987–91. https://doi.org/10.1073/pnas.0501519102 [PubMed]
- 38. Chen Y, Liu Y, Dorn GW
2nd . Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res. 2011; 109:1327–31. https://doi.org/10.1161/CIRCRESAHA.111.258723 [PubMed] - 39. Zhou H, Zhu P, Wang J, Zhu H, Ren J, Chen Y. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018; 25:1080–93. https://doi.org/10.1038/s41418-018-0086-7 [PubMed]
- 40. Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, Ahmad F, Matsui T, Chin S, Wu PH, Rybkin II, Shelton JM, Manieri M, et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005; 1:259–71. https://doi.org/10.1016/j.cmet.2005.03.002 [PubMed]
- 41. Ago T, Yeh I, Yamamoto M, Schinke-Braun M, Brown JA, Tian B, Sadoshima J. Thioredoxin1 upregulates mitochondrial proteins related to oxidative phosphorylation and TCA cycle in the heart. Antioxid Redox Signal. 2006; 8:1635–50. https://doi.org/10.1089/ars.2006.8.1635 [PubMed]
- 42. Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S. P38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004; 24:3057–67. https://doi.org/10.1128/mcb.24.7.3057-3067.2004 [PubMed]
- 43. Czubryt MP, McAnally J, Fishman GI, Olson EN. Regulation of peroxisome proliferator-activated receptor gamma coactivator 1 alpha (PGC-1 alpha) and mitochondrial function by MEF2 and HDAC5. Proc Natl Acad Sci USA. 2003; 100:1711–16. https://doi.org/10.1073/pnas.0337639100 [PubMed]
- 44. Dutta S, Sengupta P. Men and mice: relating their ages. Life Sci. 2016; 152:244–48. https://doi.org/10.1016/j.lfs.2015.10.025 [PubMed]
- 45. Ravi D, Muniyappa H, Das KC. Endogenous thioredoxin is required for redox cycling of anthracyclines and p53-dependent apoptosis in cancer cells. J Biol Chem. 2005; 280:40084–96. https://doi.org/10.1074/jbc.M507192200 [PubMed]
- 46. Bohl S, Medway DJ, Schulz-Menger J, Schneider JE, Neubauer S, Lygate CA. Refined approach for quantification of in vivo ischemia-reperfusion injury in the mouse heart. Am J Physiol Heart Circ Physiol. 2009; 297:H2054–58. https://doi.org/10.1152/ajpheart.00836.2009 [PubMed]
- 47. Das KC, Guo XL, White CW. Induction of thioredoxin and thioredoxin reductase gene expression in lungs of newborn primates by oxygen. Am J Physiol. 1999; 276:L530–39. https://doi.org/10.1152/ajplung.1999.276.3.L530 [PubMed]
- 48. Gardner PR. Aconitase: sensitive target and measure of superoxide. Methods Enzymol. 2002; 349:9–23. https://doi.org/10.1016/s0076-6879(02)49317-2 [PubMed]
- 49. Fabisiak JP, Borisenko GG, Kagan VE. Quantitative method of measuring phosphatidylserine externalization during apoptosis using electron paramagnetic resonance (EPR) spectroscopy and annexin-conjugated iron. Methods Mol Biol. 2014; 1105:613–21. https://doi.org/10.1007/978-1-62703-739-6_42 [PubMed]