



Introduction
Colon cancer (COAD) is one of the most common malignancies and the third most common cause of tumor-related mortality worldwide [1]. The survival of COAD patients highly depends on the stage of the tumor; thus, the diagnosis of COAD at an early stage can greatly enhance patients’ chances of survival [2]. The five-year survival rate is about 90% for early COAD patients, but is < 10% for advanced COAD patients [3]. Although the American Joint Committee on Cancer (AJCC) tumor, lymph node, metastases (TNM) staging system has contributed to the treatment of COAD patients, it cannot adequately predict prognosis due to the molecular heterogeneity of COAD [4]. Therefore, early diagnostic and prognostic evaluations are important to improve the treatment and overall survival (OS) of COAD patients.
Epigenetic modifications such as DNA methylation, genomic imprinting and RNA editing alter multiple signaling pathways during the development and progression of COAD [5]. The accumulation of aberrantly methylated DNA sites in intestinal epithelial cells is known to promote the occurrence of COAD [6]. DNA methylation is an important regulator of gene expression [7], and the DNA methylation status has been found to be more reliable than gene expression for the diagnosis of certain cancers [8]. DNA methylation analysis has several advantages, including a high clinical sensitivity and dynamic range, and may provide more dependable markers of COAD than gene mutation analysis [9].
Despite the benefits of DNA methylation analysis, there are limitations to the existing studies. In terms of the genome-wide DNA methylation level, non-CpG-island regions including ‘Open sea,’ ‘Shore’ and ‘Shelf’ regions account for a large proportion of total methylated positions and thus are quite likely to have important effects [10]; however, most studies have focused on abnormal DNA methylation levels in CpG islands in promoter regions. Moreover, in previous studies, methylated diagnostic biomarkers of COAD have not been able to distinguish COAD accurately and consistently from common cancers such as bladder cancer (BLCA), breast cancer (BRCA), cervical cancer (CESC), etc. For example, Sobhani et al. reported that the promoters of certain genes (including SFRP1, 2, 3, PENK, etc.) were hypermethylated in COAD, and Beggs et al. reported that five marker groups (SFRP2, SFRP4, WIF1, APC1A and APC2) could detect COAD precancerous lesions with modest predictive power (area under the curve [AUC] = 0.83), but the models in these studies could not precisely distinguish COAD from other cancers [11, 12]. Therefore, there is an urgent need for a combined diagnostic model with this ability.
Previous studies have examined not only diagnostic biomarkers, but also prognostic biomarkers of COAD. One feature of a good prognostic biomarker is its independence from clinicopathological prognostic factors. Clinicopathological characteristics such as age [13], gender [14], race [15], AJCC stage [16], examined lymph node count [17] and lymphatic invasion [18] have been identified as the primary predictors of prognosis in COAD. However, studies of methylated prognostic biomarkers thus far have not produced combined prognostic models based on genome-wide CpG sites that can effectively predict the OS of COAD patients independently of these important clinicopathological characteristics. Lind et al. reported that patients with greater methylation of a COAD biomarker group had a worse prognosis, although the difference was not dramatic in multivariate analysis [19]. Liang et al. found that methylation-regulated differentially expressed genes (5 upregulated and 81 downregulated genes) were associated with OS, but the authors did not construct a combined model to systematically predict COAD prognosis [20]. Ahn et al. demonstrated that genes such as WNT5A, SFRP1 and SFRP2 were prognostic indicators of the high CpG island methylator phenotype in COAD; however, cancer recurrence could only be predicted in resected stage III proximal COAD, not in distal COAD [21]. Thus, there is also a great need for a combined prognostic model that can accurately predict the OS of COAD patients independently of clinicopathological parameters.
A differentially methylated CpG site (DMP) is a CpG site with significantly different mean methylation levels in different groups (e.g., cancer versus normal) [22]. In this study, we used conditional screening and machine learning to obtain DMPs that could be used as specific diagnostic biomarkers for COAD. Then, we constructed and validated a COAD-specific diagnostic model using these DMPs, and evaluated its ability to distinguish COAD from normal tissues and other cancers. Finally, we constructed a combined COAD prognostic model based on six CpG sites, and verified that it could accurately predict high-risk and low-risk COAD patients independently of important clinicopathological parameters.
Results
Genomic distribution of hypermethylated and hypomethylated DMPs
To explore the abnormal methylation status of the entire genome, we conducted in-depth studies on the early diagnosis and prognostic evaluation of COAD patients (Figure 1). First, we performed CpG site expression profiling analysis between COAD tumor samples (N = 25) and paired normal samples (N = 25) from The Cancer Genome Atlas (TCGA) cohort. Then, 13716 Hyper-DMPs and 11403 Hypo-DMPs were obtained in these included cohorts. Specifically, unsupervised cluster analysis distinguished these Hyper-DMPs and Hypo-DMPs in 25 paired COAD and normal samples from TCGA (Figure 2A). When we assessed the locations of these Hyper-DMPs and Hypo-DMPs among genomic region types, we observed that Hyper-DMPs were most abundant in Island regions (39%), whereas Hypo-DMPs were mainly distributed in Open sea regions (42%) (Figure 2B). We also determined the enrichment of the DMPs by calculating the ratio of Hyper-DMPs to Hypo-DMPs in each region. The results indicated that Hyper-DMPs were enriched in Island regions (66%; Hyper/Hypo = 5421/2689), whereas Hypo-DMPs occurred more frequently in Open sea regions (58%; Hypo/Hyper = 4882/3588).
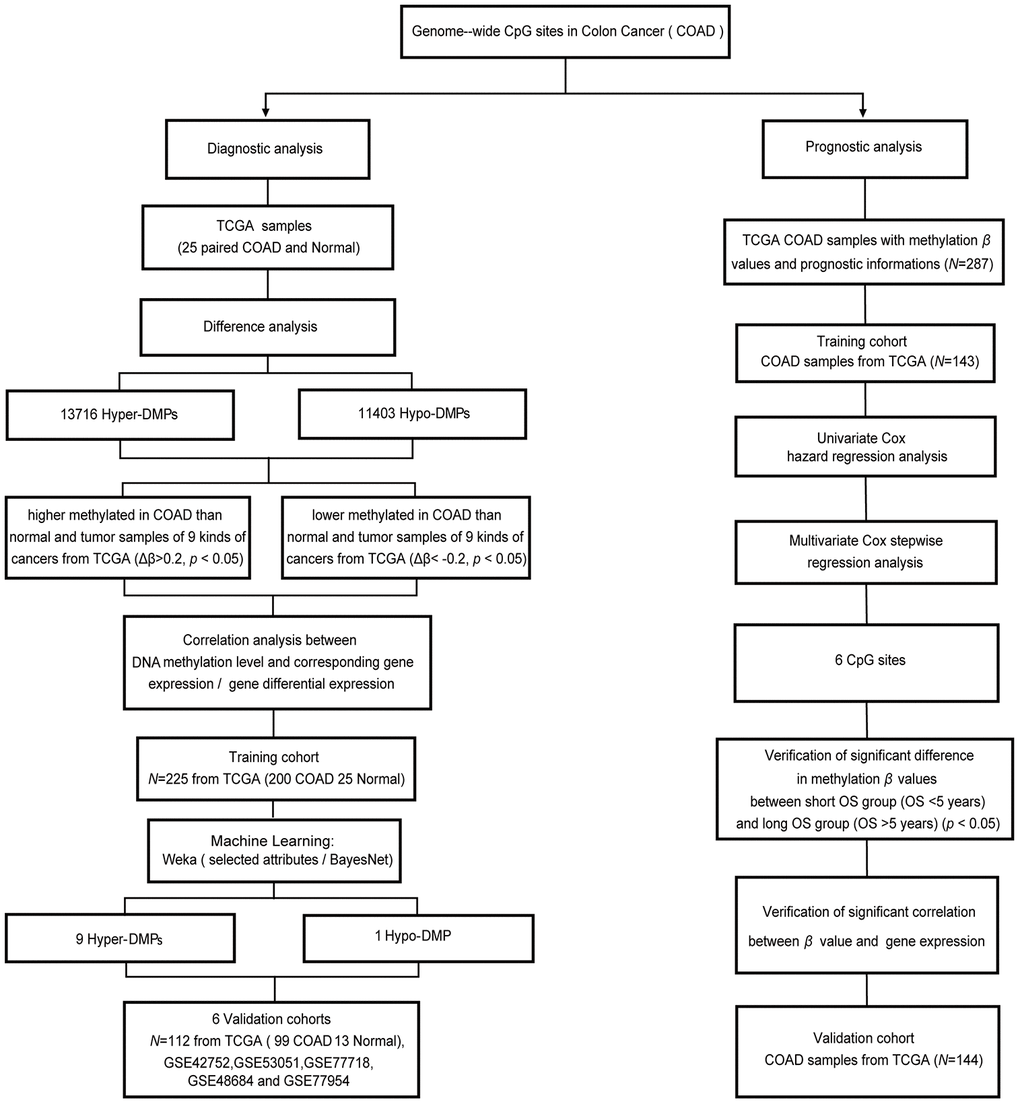
Figure 1. Workflow diagram for biomarker screening and model construction. The DNA methylation levels of genome-wide CpG sites were used to screen biomarkers and construct diagnostic and prognostic models of COAD. Left side: diagnostic biomarker selection and COAD-specific diagnostic model construction. Conditional screening and machine learning using the selected attributes and BayesNet functions of WEKA were performed to obtain the final nine Hyper-DMPs and one Hypo-DMP as potential biomarkers in the training cohort from TCGA (including 200 COAD and 25 normal samples). BayesNet was used to evaluate the COAD-specific diagnostic model based on these DMPs in the validation cohort from TCGA (including 99 COAD and 13 normal samples) and five independent GEO cohorts (GSE42752, GSE53051, GSE77718, GSE48684 and GSE77954). Right side: prognostic biomarker selection and COAD prognostic model construction. Univariate Cox hazard regression analysis and multivariate Cox stepwise regression analysis were applied to 143 TCGA COAD samples as the training cohort to obtain six CpG sites as potential biomarkers. The prognostic model based on these six CpG sites was evaluated using 144 TCGA COAD samples as the validation cohort.
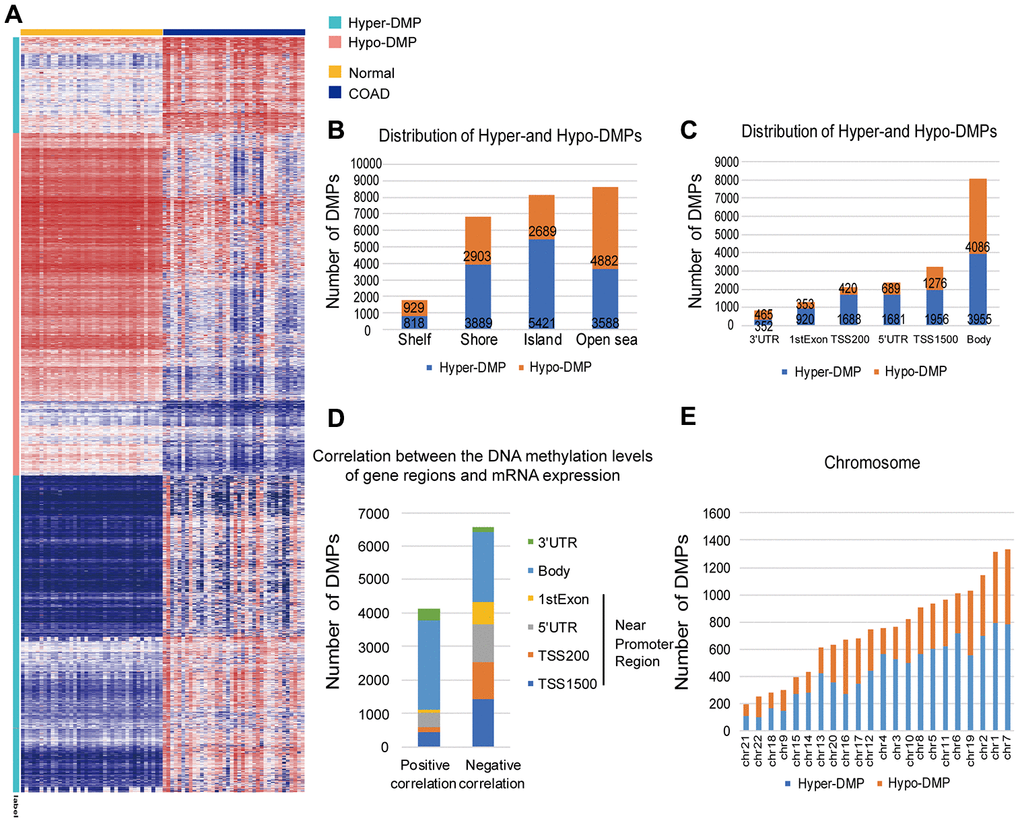
Figure 2. Distribution of DMPs. (A) Unsupervised hierarchical clustering and heat map display of the methylation levels of the Hyper- and Hypo-DMPs in 25 paired COAD and normal samples from TCGA. (B) The distribution of Hyper-DMPs and Hypo-DMPs in different genomic region types. Island, a CpG site located within a CpG island; Shore, a CpG site located < 2 kilobases from a CpG island; Shelf, a CpG site located > 2 kilobases from a CpG island; Open sea, a CpG site not in an island or annotated gene. (C) The numbers and ratios of Hyper-DMPs and Hypo-DMPs according to their distance from the promoter. TSS1500, 200-1500 base pairs upstream of the transcription start site; TSS200, 200 base pairs upstream of the transcription start site; 5′UTR, 5′ untranslated region; 1st Exon, exon 1; 3′UTR, 3′ untranslated region. (D) The positional distribution (in terms of promoter distance) of the DMPs in which the methylation level correlated positively or negatively with the expression of the corresponding gene (FDR < 0.05). (E) Chromosome distribution of Hyper-DMPs and Hypo-DMPs. Chr: chromosome.
More importantly, Hyper-DMPs were mainly located near promoter regions, including TSS1500 (the region 200 to 1500 nucleotides upstream of the transcription start site), TSS200 (the region from the transcription start site to 200 nucleotides upstream of the transcription start site), the 5′ untranslated region (UTR) and the 1st Exon (Figure 2C). However, Hypo-DMPs were mostly enriched in the Body and the 3′UTR, which occupied a large percentage of the regions, genome-wide. The DMP distribution ratio also indicated that proximal promoter regions were mainly hypermethylated (69%; hyper/hypo = 6245/2738), while the proportions of Hyper- and Hypo-DMPs in the Body and 3′UTR were almost equal (51%; hypo/hyper = 4551/4307). Notably, both Hyper-DMPs and Hypo-DMPs occupied a large proportion of the whole genome, about 3.42%.
Next, we calculated Pearson correlation coefficients to determine the correlation between the DNA methylation of the DMPs and the expression of their corresponding genes (Figure 2D). Among the 17112 DMPs for which both the DNA methylation levels and the corresponding mRNA expression profiles were available, the methylation levels of 6565 Hyper-DMPs and 4112 Hypo-DMPs were significantly associated with the mRNA levels of the corresponding genes (|r| > 0.1, false discovery rate [FDR] < 0.05). When we analyzed the distance between these DMPs and promoter regions, we found that DMPs in or near promoter regions (i.e., in the 1st Exon, 5′ UTR, TSS200 or TSS1500) were negatively associated with mRNA expression, whereas those outside promoter regions (i.e., in the Body or 3′ UTR) were positively associated with gene expression. Moreover, the DMPs had a higher distribution frequency on chromosomes 7 and 1 than on the other autosomes (Figure 2E). Since the DMPs that significantly altered the expression of their corresponding genes were not limited to promoter regions, we screened the whole genome for potential biomarkers of COAD and constructed a diagnostic prediction model based on the genome-wide Hyper- and Hypo-DMPs.
Identification of COAD-specific methylation biomarkers and construction of a COAD-specific diagnostic model
Next, in order to construct a diagnostic model to distinguish COAD tumor tissues from normal intestinal epithelial tissues and the tumor tissues of nine other cancer types (BLCA, BRCA, CESC, glioblastoma [GBM], head and neck cancer [HNSC], liver cancer [LIHC], lung adenocarcinoma [LUAD], lung squamous cell carcinoma [LUSC] and endometrial cancer [UCEC]), we performed conditional screening and machine learning studies based on the genome-wide DMPs obtained from TCGA above (13716 Hyper-DMPs and 11403 Hypo-DMPs). For the conditional screening, we determined the average β values (a measure of CpG site methylation) of these Hyper-/Hypo-DMPs in all the samples for the nine cancer types in TCGA. Then, we selected DMPs based on an average methylation level difference of at least 0.2 units in COAD. After screening under the above conditions, we obtained 17 Hyper-DMPs and 8 Hypo-DMPs as candidate biomarkers. Further analysis revealed that these 17 Hyper-DMPs and 8 Hypo-DMPs were among the 6565 Hyper-DMPs and 4112 Hypo-DMPs that were significantly associated with the expression of their corresponding genes (log2 |fold change| > 1, FDR < 0.05).
For machine learning, two-thirds of the total tumor and normal samples from the COAD cohort of TCGA (200 COAD and 25 normal samples) were randomly set as the training cohort, while the remaining one-third of the total samples (99 COAD and 13 normal samples) were used as the validation cohort. The β values of the 17 Hyper-DMPs and 8 Hypo-DMPs in the training cohort were input into WEKA, and the selected attributes function of WEKA was used to filter these candidate biomarkers. As potential diagnostic biomarkers, nine Hyper-DMPs (cg26036626, cg03882585, cg08130988, cg16733654, cg12587766, cg08808128, cg13004587, cg05038216 and cg09746736) and one Hypo-DMP (cg26718707) were selected to construct a COAD-specific diagnostic model (Table 1). Finally, based on the nine Hyper-DMPs and one Hypo-DMP, we constructed a COAD-specific diagnostic model with BayesNet [23].
Table 1. Characteristics of 9 Hyper-DMPs and 1 Hypo-DMP in the COAD-specific diagnostic model.
| Biomarkers | Ref Gene | Chromosome | Start | End | CGI Coordinate | Feature | CGI | FDR | Type | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg26036626 | FBLIM1 | chr1 | 15759102 | 15759103 | 15758576-15759367 | 5'UTR | Island | 1.79e-15 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg03882585 | SYNE1 | chr6 | 152636775 | 152636776 | 152636675-152637337 | 5'UTR | Island | 1.29e-05 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg08130988 | EFEMP1 | chr2 | 55923790 | 55923791 | 55923205-55923813 | 1st Exon | Island | 6.49e-05 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg16733654 | PTPRS | chr19 | 5293072 | 5293073 | 5292760-5294200 | 5'UTR | Island | 7.65e-09 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg12587766 | LIFR | chr5 | 38556333 | 38556334 | 38556120-38557461 | 1st Exon | Island | 2.19e-11 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg08808128 | CLIP4 | chr2 | 29115566 | 29115567 | 29115117-29116043 | 1st Exon | Island | 4.48e-09 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg13004587 | SCGB3A1 | chr5 | 180590349 | 180590350 | 180590099-180592062 | Body | Island | 0.0448 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg05038216 | CLIP4 | chr2 | 29116225 | 29116226 | 29115117-29116043 | 5'UTR | Shore | 1.82e-09 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg09746736 | SLC6A2 | chr16 | 55656218 | 55656219 | 55655686-55656983 | TSS 1500 | Island | 2.38e-08 | Hyper-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg26718707 | DIP2C | chr10 | 472430 | 472431 | 472252-472531 | Body | Island | 0.0107 | Hypo-DMP | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| CGI: CpG island | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The average β values of the nine Hyper-DMPs and one Hypo-DMP selected for our diagnostic model in all the COAD tissues, normal tissues and nine types of cancerous tissues from TCGA are visualized in Figure 3A. We performed an unsupervised cluster analysis to evaluate these β values (Figure 3B), and found that they were clearly divided into four clusters. The COAD tumor samples were significantly differentiated from all the normal samples and the tumor samples from the nine other cancer types based on the nine Hyper-DMPs and one Hypo-DMP.
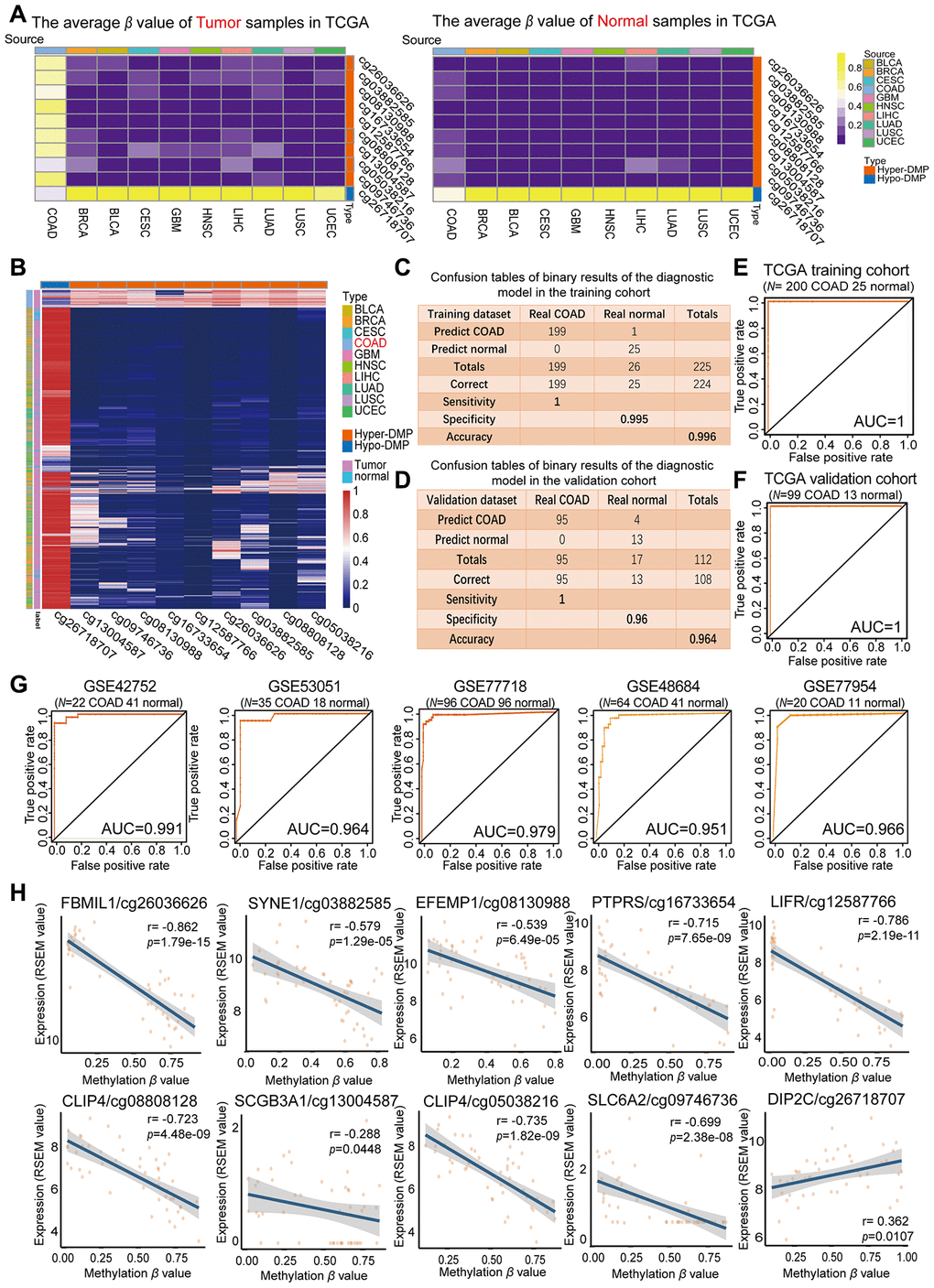
Figure 3. Evaluation of the COAD-specific diagnostic biomarkers and diagnostic model. (A) Heat maps of the average methylation levels of the nine Hyper-DMPs and one Hypo-DMP in all the samples from 10 cancer types. The legend on the right marks the source and CpG type. The picture on the left represents the tumor samples in TCGA, while the picture on the right represents the normal samples in TCGA. (B) Unsupervised hierarchical clustering of the methylation levels of the nine Hyper-DMPs and one Hypo-DMP in all the samples from 10 cancer types. The legend on the right marks the source and CpG type. (C–F) Confusion tables (C, E) and corresponding ROC curves (D, F) for the binary results of the COAD-specific diagnostic model in the training cohort (N = 225) and the validation cohort (N = 112) from TCGA. (G) ROC curves of the COAD-specific diagnostic model in five GEO COAD validation cohorts (GSE42752, GSE53051, GSE77718, GSE48684 and GSE77954, which included 22 COAD and 41 normal samples, 35 COAD and 18 normal samples, 96 paired COAD and normal samples, 64 COAD and 41 normal samples, and 20 COAD and 11 normal samples, respectively). (H) The correlation between the DMP methylation level and the expression of the corresponding gene for each diagnostic biomarker, determined through Pearson correlation tests (r > 0.2, FDR < 0.05). Gene expression is presented as the RSEM normalized count converted by log2 (x + 1).
Subsequently, we used the COAD-specific diagnostic model to train the training cohort (including 200 COAD and 25 normal samples from TCGA) in WEKA. Using BayesNet, we determined that the COAD-specific diagnostic model had a sensitivity of 100%, specificity of 99.5% and accuracy of 99.6% in the training cohort (Figure 3C). In the validation cohort (99 COAD and 13 normal samples from TCGA), the diagnostic model had a sensitivity of 100%, specificity of 96% and accuracy of 96.4% (Figure 3D). Therefore, our COAD-specific diagnostic model was confirmed to perfectly distinguish between COAD and normal samples in the training cohort (AUC = 1) (Figure 3E) and the validation cohort (AUC = 1) from TCGA (Figure 3F).
To demonstrate the versatility of our diagnostic model, we conducted a population heterogeneity analysis using the validation cohort from TCGA, which included samples from 4 Asian, 25 black or African American and 73 white patients. The sensitivity, specificity and accuracy are shown in Table 2. Our diagnostic model exhibited no significant population heterogeneity, suggesting that it can be applied to people of different races. In addition, we used the five independent GEO COAD cohorts mentioned above (GSE42752, GSE53051, GSE77718, GSE48684 and GSE77954) as validation cohorts. In receiver operating characteristic (ROC) analyses, the AUCs of these five cohorts were 0.991, 0.964, 0.979, 0.951 and 0.966, respectively (Figure 3G). These results further illustrated the reproducibility and stability of our COAD-specific diagnostic model.
Table 2. The stratification analysis of the sensitivity, specificity, and accuracy from different races including 4 Asian, 25 Black or African American, and 73 White upon TCGA validation cohort.
| Race | Asian | Black or African American | White |
| Sensitivity | 0 | 1 | 1 |
| Specificity | 1 | 0.913 | 0.97 |
| Accuracy | 1 | 0.92 | 0.973 |
We also analyzed the correlation between the DMP methylation level and the expression of the corresponding genes for the nine Hyper-DMPs and the one Hypo-DMP in our diagnostic model. The Hypo-DMP (cg26718707) corresponded to DIP2C, while the nine Hyper-DMPs corresponded to eight genes: FBLIM1 (cg26036626), SYNE1 (cg03882585), EFEMP1 (cg08130988), PTPRS (cg16733654), LIFR (cg12587766), CLIP4 (cg08808128 and cg05038216), SCGB3A1 (cg13004587) and SLC6A2 (cg09746736). The results of the correlation analysis are shown in Figure 3H and Supplementary Table 1. The expression of DIP2C correlated positively with the methylation level of the Hypo-DMP (r > 0.1, FDR < 0.05), and the expression of the other eight genes correlated negatively with the methylation levels of the corresponding nine Hyper-DMPs (r < -0.1, FDR < 0.05).
Next, using the five independent GEO cohorts, we compared our COAD-specific diagnostic model with three previously reported methylation-based diagnostic models: a Bayesian model including four CpG sites from Azuara et al. [24], a logistic regression model including five CpG sites from Beggs et al. [25] and a logistic regression model including 12 CpG sites from Naumov et al. [26] (Figure 4A). As expected, our model exhibited better sensitivity, specificity and accuracy than the three previously reported diagnostic models in most cases for the five GEO COAD cohorts. We also compared our diagnostic model with these three models in terms of its ability to distinguish COAD from normal tissues and nine other types of cancerous tissues. For this purpose, we divided all the samples from the five GEO COAD cohorts and nine various TCGA cancerous cohorts into a tumor group and a normal group, and we calculated the proportion of samples that were predicted to be COAD among all the samples (Figure 4B, Table 3). In the cohorts from the nine different types of cancers, the ideal proportion would have been 0. When we tested our COAD-specific diagnostic model, almost none of the normal intestinal epithelial samples or the tumor tissues from the nine other cancer types were predicted as COAD (0-5%, median: 0%). However, when we tested the three previously reported diagnostic models, 0-97.7% of the normal tissues (median: 0%, 33.3% and 0%, respectively) and 20.2-98.7% of the tumor tissues from the nine other cancer types (median: 45.4%, 93.5% and 75.2%, respectively) were predicted as COAD. Therefore, our COAD-specific diagnostic model based on nine Hyper-DMPs and one Hypo-DMP not only distinguished COAD from normal samples, but also compensated for the deficiencies of previous COAD diagnostic models that could not differentiate COAD from nine other cancer types.
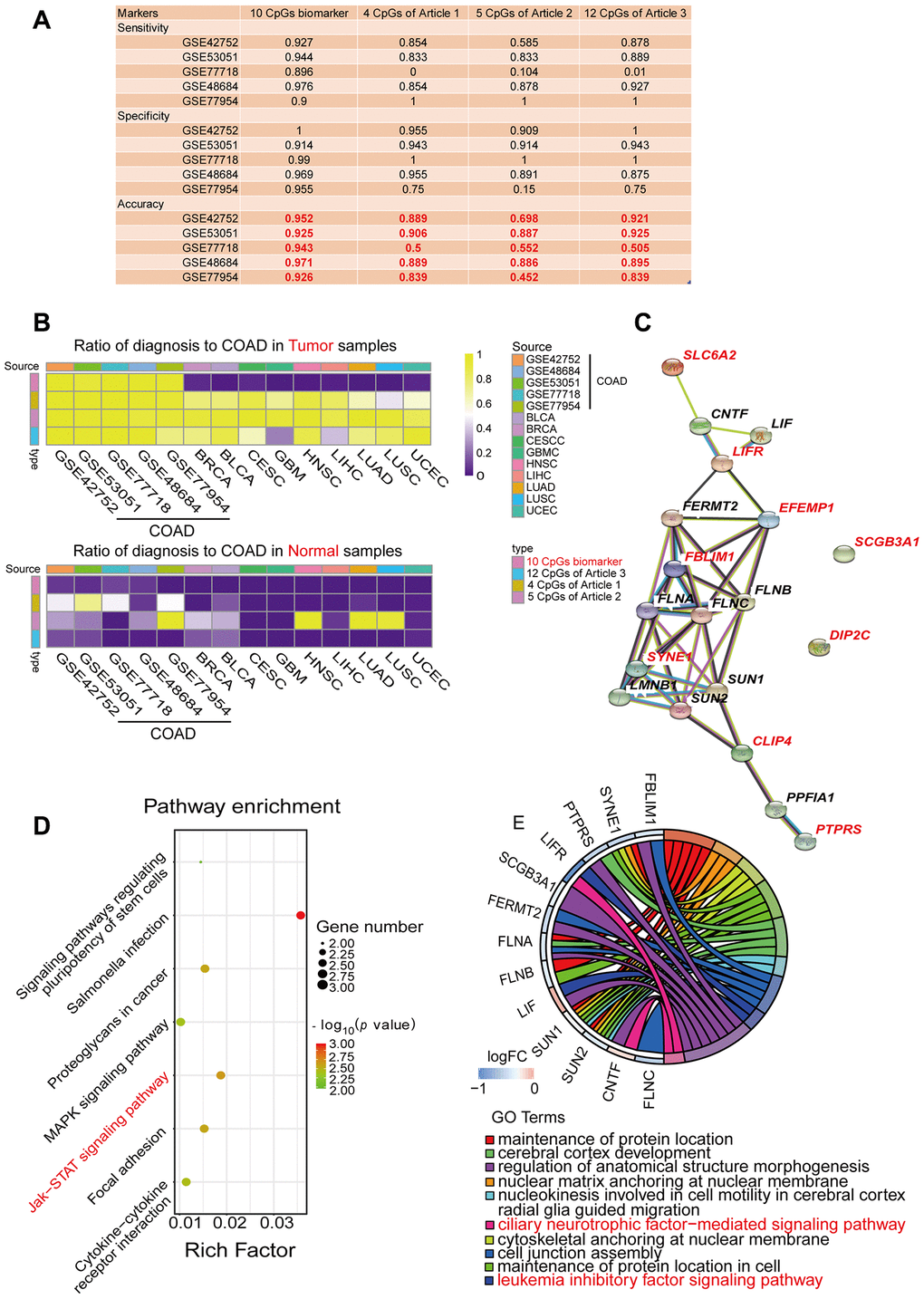
Figure 4. Performance comparison of diagnostic models and enrichment analysis of the corresponding genes. (A) Table displaying the classification performance of different methylation models for COAD and normal tissues in five independent GEO cohorts (GSE42752, GSE53051, GSE77718, GSE48684 and GSE77954). In addition, Azuara et al. [24] (Article 1) reported four CpG sites as diagnostic biomarkers for COAD, and the methylation values for each of them were available in the COAD cohort from TCGA; Beggs et al. [25] (Article 2) reported six CpG sites as diagnostic biomarkers for COAD, and the methylation values for five of them were available in the COAD cohort from TCGA; and Naumov et al. [26] (Article 3) reported 14 CpG sites as diagnostic biomarkers for COAD, and the methylation values for 12 of them were available in the COAD cohort from TCGA. (B) Heat map comparing our diagnostic model with the previous methylation models. Rows are labeled with the different sources of methylation data. The legend indicates that the range is 0-1. The color represents the percentage of the total samples predicted to be COAD. In the cohorts for the nine different cancer types, the ideal results should be 0. (C) Predicted protein interaction network of the genes corresponding to the COAD-specific diagnostic biomarkers. Version 11.0 of the STRING protein database was used. The different line colors represent different kinds of correlations between the proteins corresponding to the model (dark blue for coexistence, black for co-expression, pink for an experiment, light blue for a database, green for text mining, and purple for homology). The red genes are the corresponding genes of the diagnostic biomarkers. Note that CLIP4 is the corresponding gene for both cg08808128 and cg05038216. (D, E) KEGG (D) and GO (E) enrichment analysis results from the STRING protein database. All seven results are shown for the KEGG enrichment analysis, and the top 10 results are shown for the GO enrichment analysis, with p-values arranged from large to small. In the KEGG enrichment graph (D), the X-axis represents the Rich factor, indicating the degree of enrichment (Rich factor = observed gene counts/background gene counts), and the Y-axis represents the enriched KEGG terms. The color represents the -log10 (p-value), and the size of the dot represents the number of genes. In the GO enrichment graph (E), the GO term indicates the GO enrichment pathway.
Table 3. Comparison of the performance of different methylation models in all samples of 9 kinds of cancers.
| Accuracy | BLCAC | BLCAN | BRCAC | BRCAN | CESCC | CESCN | GBMC | GBMN | HNSCC | HNSCN | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 DMPs biomarker | 0.022 | 0 | 0.05 | 0 | 0.05 | 0 | 0 | 0 | 0.02 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 CpGs of Article 1 | 0.593 | 0.095 | 0.584 | 0.01 | 0.663 | 0 | 0.327 | 0 | 0.742 | 0.02 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 CpGs of Article 2 | 0.939 | 0.333 | 0.92 | 0.388 | 0.977 | 0 | 0.928 | 0 | 0.977 | 0.977 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 CpGs of Article 3 | 0.872 | 0.19 | 0.749 | 0.143 | 0.625 | 0 | 0.242 | 0 | 0.87 | 0.02 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accuracy | LIHCC | LIHCN | LUADC | LUADN | LUSCC | LUSCN | UCECC | UCECN | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 10 DMPs biomarker | 0.03 | 0 | 0.01 | 0 | 0 | 0 | 0.04 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 4 CpGs of Article 1 | 0.413 | 0 | 0.454 | 0 | 0.202 | 0 | 0.348 | 0 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 5 CpGs of Article 2 | 0.871 | 0.08 | 0.987 | 1 | 0.935 | 0.93 | 0.843 | 0.043 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| 12 CpGs of Article 3 | 0.375 | 0 | 0.752 | 0 | 0.815 | 0 | 0.944 | 0.043 | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| The vertical axis shows the different sources of the methylation models. The horizontal axis shows the tumor and normal methylation cohorts from 9 kinds of cancer. Affix C represents the tumor samples of the methylation data sets. Affix N represents the normal sample of the methylation cohorts. The numbers represent the percentage of samples predicted to be COAD in the total samples. The cohorts in the table are from 9 kinds of cancers, and the ideal result should be 0. Azuara D et al. [24] (Article 1)reported 4 CpG sites as diagnostic markers for COAD, and 4 of them had methylation values in the TCGA COAD cohort; Beggs AD et al. [25] (Article 2)report 6 CpG sites as diagnostic markers for COAD, and 5 of them had methylation values in the TCGA COAD cohort; Naumov VA et al. [26] (Article 3)reported 14 CpG sites as diagnostic markers for COAD, and 12 of them had methylation values in the TCGA COAD cohort. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Then, we used the STRING database to construct a protein-protein interaction network for the nine genes corresponding to the nine Hyper-DMPs and the one Hypo-DMP (Figure 4C). We also performed Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses on these genes (Figure 4D and 4E). We found that the nine genes were involved in important signaling pathways of tumorigenesis and development, such as Salmonella infection, Janus kinase (JAK)/signal transducer and activator of transcription (STAT) signaling, focal adhesion, proteoglycans in cancer, cytokine-cytokine receptor interactions, etc. All the results of the KEGG pathway analysis and the top 10 results of the GO analysis are shown in Supplementary Tables 2 and 3.
The above results demonstrated that our COAD-specific diagnostic model could accurately and precisely distinguish COAD tissues from normal intestinal epithelial samples and tumor samples from nine cancer types, and that the nine Hyper-DMPs and one Hypo-DMP included in this model may be potential biomarkers for the early prediction and specific diagnosis of COAD.
Identification of prognostic biomarkers of COAD and construction of a combined COAD prognostic model
A total of 287 COAD tissue samples in the cohort from TCGA had both methylated β values and corresponding prognostic information. The distribution and corresponding demographic characteristics of these patients are summarized in Table 4. The patients were divided into a training cohort (N = 143) and a validation cohort (N = 144). The training cohort was used to obtain prognostic biomarkers and construct a COAD prognostic model, while the validation cohort was used to test the COAD prognostic model. Univariate Cox hazard regression analysis of the training cohort revealed 64 CpG sites that correlated significantly with the OS of COAD patients (FDR < 0.05); thus, these CpG sites were identified as candidate prognostic biomarkers. Multivariate Cox stepwise regression analysis was applied to these 64 CpG sites, and six sites (cg00177496, cg01963906, cg05165940, cg12921795, cg19414598 and cg25783173) were included in our final hazard ratio model, which was constructed as a combined COAD prognostic model for OS prediction (Table 5). The six CpG sites from our COAD prognostic model were found to correspond to BDH1 (cg00177496), SYTL1 (cg01963906), SATB2 (cg05165940), WDR20 (cg12921795), DMC1 (cg19414598) and ZNF35 (cg25783173) (Figure 5A and Supplementary Table 4). The expression of SYTL1 correlated positively with the methylation level of cg01963906 (r > 0.1, FDR < 0.05), and the expression of the other five genes correlated negatively with the methylation levels of the remaining CpG sites (r < -0.1, FDR < 0.05).
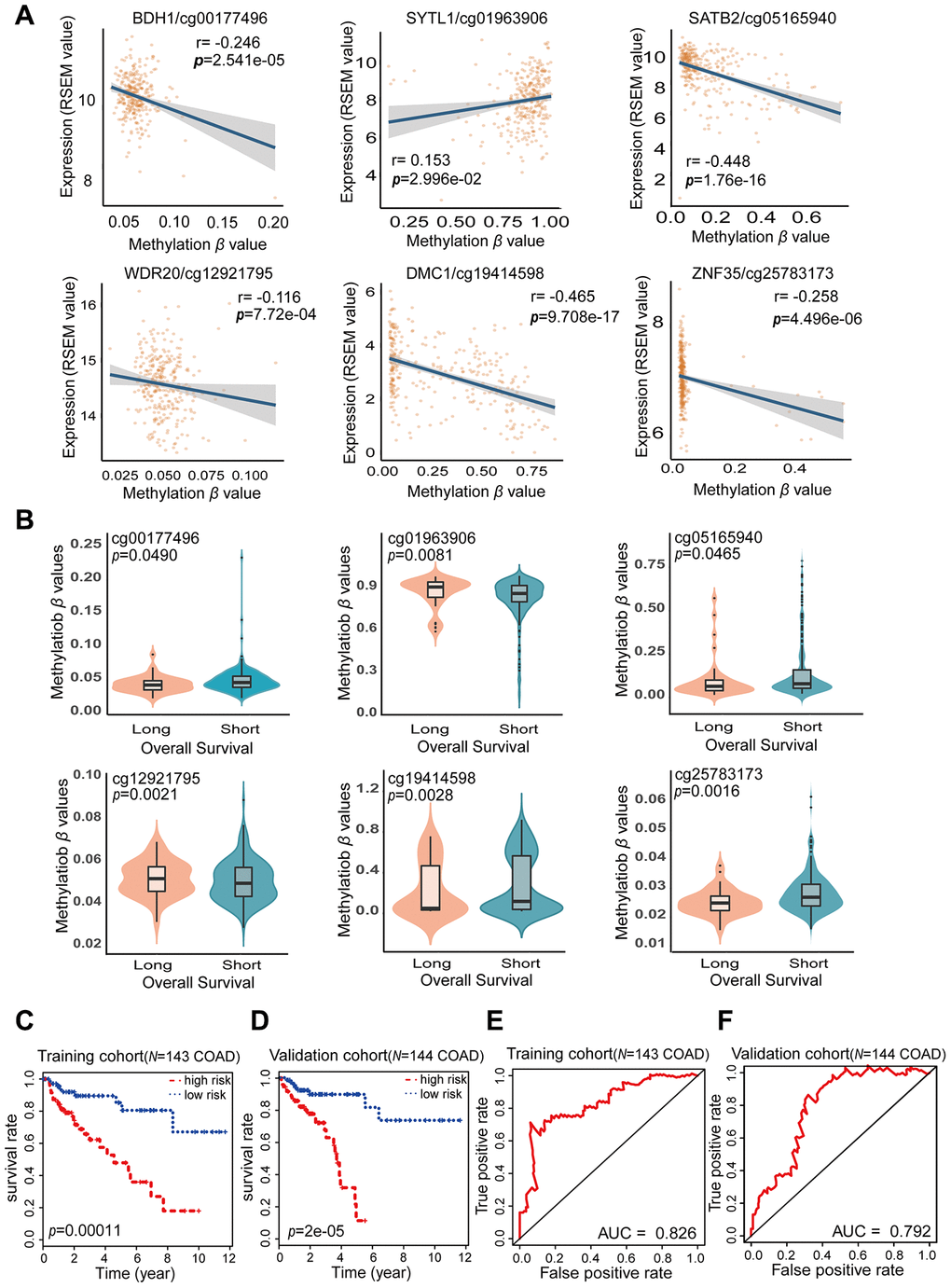
Figure 5. Characteristics of the potential prognostic biomarkers and evaluation of the combined prognostic model based on six CpG sites. (A) The correlations between the methylation β levels of the prognostic biomarkers and the expression of the corresponding genes were evaluated with Pearson correlation tests. Gene expression is presented as the RSEM normalized count converted by log2 (x + 1). (B) Violin plots of the methylation β values for patients with longer (> 5 years) and shorter (< 5 years) OS in the training cohort, with the median in the centerline. A Wilcoxon test was used to determine the difference between the two groups. The corresponding CpG sites, cor-values and p-values are shown at the top of the plot. (C, D) Kaplan-Meier analysis was performed on the OS of high-risk and low-risk patients using our prognostic model in the training (N = 143) (C) and validation (N = 144) (D) cohorts from TCGA. The difference in OS between the two groups was determined with a log-rank test. Higher risk scores were associated with significantly poorer OS. Patients were divided into low-risk and high-risk groups using the median risk score as the cut-off. (E, F) ROC curves showing the sensitivity and specificity of the prognostic model in predicting patients’ OS in the training (N = 143) (E) and validation (N = 144) (F) cohorts from TCGA.
Table 4. Clinicopathological characteristics of COAD patients from the TCGA database.
| Characteristics | Patients | |||||
| Total (N = 287) | Training cohort (N = 143) | Validation cohort (N = 144) | ||||
| No | % | No | % | No | % | |
| Age | ||||||
| ≤64 | 130 | 45.30 | 58 | 40.56 | 72 | 50 |
| >64 | 157 | 54.70 | 85 | 59.44 | 72 | 50 |
| Histological type | ||||||
| Colon Adenocarcinoma | 246 | 85.71 | 119 | 83.22 | 127 | 88.19 |
| Colon Mucinous Adenocarcinoma | 38 | 13.24 | 22 | 15.38 | 16 | 11.11 |
| Unknown | 3 | 1.05 | 2 | 1.40 | 1 | 0.70 |
| Pathologic M | ||||||
| M0 | 195 | 67.94 | 102 | 71.33 | 93 | 64.58 |
| M1 | 40 | 13.94 | 21 | 14.69 | 19 | 13.19 |
| MX | 49 | 17.07 | 19 | 13.29 | 30 | 20.83 |
| Unknown | 3 | 1.05 | 1 | 0.07 | 2 | 1.39 |
| Pathologic N | ||||||
| N0 | 166 | 57.84 | 73 | 51.05 | 93 | 64.58 |
| N1 | 73 | 25.44 | 46 | 32.17 | 27 | 18.75 |
| N2 | 48 | 16.72 | 24 | 1.40 | 24 | 16.67 |
| Pathologic T | ||||||
| T1 | 7 | 2.44 | 2 | 1.39 | 5 | 3.47 |
| T2 | 42 | 14.63 | 17 | 11.89 | 25 | 17.36 |
| T3 | 199 | 69.34 | 105 | 73.43 | 94 | 65.28 |
| T4 | 38 | 13.24 | 19 | 13.29 | 19 | 13.19 |
| Unknown | 1 | 0.35 | 0 | 0 | 1 | 0.70 |
| Gender | ||||||
| Female | 134 | 46.69 | 70 | 48.95 | 64 | 44.44 |
| Male | 153 | 53.31 | 73 | 51.05 | 80 | 55.56 |
| Race | ||||||
| American Indian or Alaska Native | 1 | 0.35 | 1 | 0.70 | 0 | 0 |
| Asian | 11 | 3.83 | 4 | 2.80 | 7 | 4.86 |
| Black or African American | 57 | 19.86 | 20 | 13.98 | 37 | 25.70 |
| White | 201 | 70.04 | 108 | 75.52 | 93 | 64.58 |
| Unknown | 17 | 5.92 | 10 | 7.00 | 7 | 4.86 |
| Tumor stage | ||||||
| Stage I | 43 | 14.98 | 16 | 11.19 | 27 | 18.75 |
| Stage II | 110 | 38.33 | 52 | 36.36 | 58 | 40.28 |
| Stage III | 84 | 29.27 | 51 | 14.69 | 33 | 22.92 |
| Stage IV | 40 | 13.94 | 21 | 2.10 | 19 | 13.19 |
| Unknown | 10 | 3.48 | 3 | 27.27 | 7 | 4.86 |
| Lymphatic invasion | ||||||
| Yes | 76 | 26.48 | 39 | 27.27 | 37 | 25.69 |
| No | 175 | 60.98 | 88 | 61.54 | 87 | 60.42 |
| Unknown | 36 | 12.54 | 16 | 11.19 | 20 | 13.89 |
| Primary lymph node presentation assessment | ||||||
| Yes | 265 | 92.33 | 134 | 93.70 | 131 | 90.97 |
| No | 14 | 4.88 | 5 | 3.50 | 9 | 6.25 |
| Unknown | 8 | 2.79 | 4 | 2.80 | 4 | 2.78 |
| Vital status | ||||||
| Alive | 218 | 75.96 | 107 | 74.83 | 111 | 77.08 |
| Dead | 69 | 24.04 | 36 | 25.17 | 33 | 22.92 |
| Longest dimension | ||||||
| ≥2 | 43 | 14.98 | 27 | 18.88 | 16 | 11.11 |
| <2 | 175 | 60.98 | 90 | 62.94 | 85 | 59.03 |
| Unknown | 69 | 24.04 | 26 | 18.18 | 43 | 29.86 |
| Sample type | ||||||
| Metastatic | 1 | 0.35 | 0 | 0 | 1 | 0.69 |
| Primary Tumor | 285 | 99.30 | 142 | 99.30 | 143 | 99.31 |
| Recurrent Tumor | 1 | 0.35 | 1 | 0.70 | 0 | 0 |
| Lymph node examined count | ||||||
| ≥12 | 226 | 78.75 | 117 | 81.82 | 109 | 75.70 |
| <12 | 42 | 14.63 | 19 | 13.28 | 23 | 15.97 |
| Unknown | 19 | 6.62 | 7 | 4.90 | 12 | 8.33 |
Table 5. Characteristics of prognostic biomarkers and their coefficients in the combined COAD prognostic model.
| Biomarkers | Ref Gene | Coefficients | HR | CI (lower) | CI (upper) | SE | z value | CGI | FDR | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg00177496 | BDH1 | 38.52 | 5.3E+16 | 3.55E+02 | 8.02E+30 | 16.66 | 2.312 | Island | 2.541e-05 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg01963906 | SYTL1 | -4.13 | 0.01608 | 1.86E-03 | 1.39E-01 | 1.102 | -3.75 | Island | 2.996e-02 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg05165940 | SATB2 | 2.574 | 13.12 | 2.70E+00 | 6.38E+01 | 0.8072 | 3.189 | Island | 1.76e-16 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg12921795 | WDR20 | -79.32 | 3.58E-35 | 2.16E-57 | 5.91E-13 | 26.1 | -3.039 | Island | 7.72e-04 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg19414598 | DMC1 | 2.31 | 10.07 | 2.75E+00 | 3.68E+01 | 0.6616 | 3.491 | Island | 9.708e-17 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| cg25783173 | ZNF35 | 6.061 | 429 | 1.60E+01 | 1.15E+04 | 1.677 | 3.614 | Island | 4.496e-06 | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| HR: Hazard Ratio; CI: 95.0% confidence interval; SE: standard errors of coefficients; z value: Wald z-statistic value. | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
The risk score formula for our COAD prognostic model was based on the regression coefficients and methylation levels of the six CpG sites, as follows: risk score = (38.52 × cg00177496 β value) – (4.13 × cg01963906 β value) + (2.574 × cg05165940 β value) – (79.32 × cg12921795 β value) + (2.31 × cg19414598 β value) + (6.061 × cg25783173 β value). In the risk score formula, a positive coefficient for a CpG site (cg00177496, cg05165940, cg19414598 and cg25783173) indicates that hypermethylation of that site was associated with shorter OS in COAD patients. In contrast, a negative coefficient for a CpG site (cg01963906 and cg12921795) indicates that greater methylation of that site was associated with longer OS. Our COAD prognostic model revealed that there were significant differences in DNA methylation levels between patients with long-term (> 5 years) and short-term (< 5 years) survival (FDR < 0.05) (Figure 5B). Consistent with the results of the multivariate Cox stepwise regression analysis, the CpG sites with positive coefficients (cg00177496, cg05165940, cg19414598 and cg25783173) exhibited lower methylation levels in patients who survived long-term, while the CpG sites with negative coefficients (cg01963906 and cg12921795) exhibited higher methylation levels in patients who survived long-term. Thus, our combined COAD prognostic model based on six CpG sites successfully distinguished long-term from short-term surviving patients in the training cohort of 143 COAD samples from TCGA.
Based on the Cox regression analyses, risk scores were used as continuous variables in the training and validation cohorts. The risk scores obtained from the combined COAD prognostic model correlated significantly with the OS of COAD patients (Training cohort: likelihood ratio test = 45, p < 0.0001; Wald test = 41.93, p < 0.0001; score [log-rank] test = 50.34, p < 0.0001. Validation cohort: likelihood ratio test = 27.63, p < 0.0001; Wald test = 33.48, p < 0.0001; score [log-rank] test = 39.33, p < 0.0001). Using the median risk score of the training cohort as a cut-off value, we generated Kaplan-Meier curves and performed log-rank tests on the training cohort (Figure 5C) (p = 0.00011) and the validation cohort (Figure 5D) (p = 2e-05). Through these analyses, we sought to compare the OS of patients in the high-risk and low-risk groups and thus determine the predictive value of the combined COAD prognostic model based on six CpG sites. The risk scores for the training and validation cohorts are shown in Supplementary Tables 5 and 6. The survival rate of patients was significantly greater in the low-risk group than in the high-risk group. These results confirmed that our combined prognostic model based on six CpG sites could classify patients into high-risk and low-risk groups, demonstrating its clinical practicability.
To further evaluate the specificity of our combined COAD prognostic model in predicting survival, we used the AUC values obtained from time-dependent ROC analyses as categorical variables. In both the training cohort and the validation cohort, the combined COAD prognostic model precisely predicted the survival of COAD patients, with AUC values of 0.826 and 0.792, respectively (Figure 5E and 5F). We also performed univariate Cox regression analyses of the six individual CpG sites included in the COAD prognostic model (Supplementary Figure 1). The calculated AUC values indicated that the six individual CpG sites could also distinguish high-risk from low-risk patients; however, the predictive effect of any one CpG site was not as good as the predictive effect of the combined prognostic model using all six CpG sites. These results demonstrated that the six CpG sites may be potential prognostic biomarkers of COAD, but the combined COAD prognostic model based on six CpG sites is more valuable than the individual sites for clinical validation and prognostic evaluation.
Independence of the combined COAD prognostic model in OS prediction, and comparison of our prognostic model with other known prognostic models
To evaluate the stability and independence of our combined COAD prognostic model based on six CpG sites, we stratified patients according to clinicopathological characteristics such as age, gender, race, AJCC stage, examined lymph node count and lymphatic invasion. Remarkably, the Kaplan-Meier plots displayed significant extension of OS in the low-risk groups for all these characteristics in the 287 COAD samples from TCGA. Nevertheless, the combined COAD prognostic model predicted the survival of COAD patients more precisely than these factors, with an AUC value of 0.687 (Figure 6A–6C, Figure 7A–7C and Supplementary Figure 2). These results confirmed that the combined COAD prognostic model based on six CpG sites provided an excellent reference for different populations and was an independent predictor of patient survival.
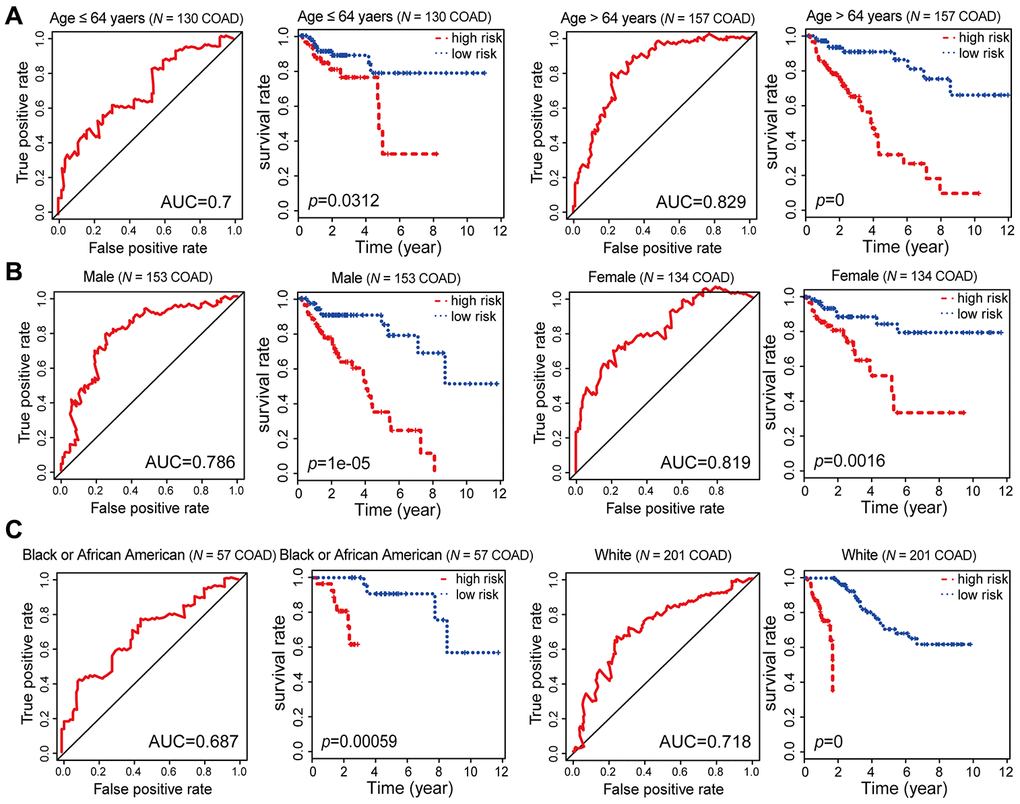
Figure 6. Kaplan-Meier and ROC analysis results based on age, gender and race. (A) Grouping of COAD patients according to their age at first diagnosis: ≤ 64 years (N = 130, 45.30%), > 64 years (N = 157, 54.70%). (B) Grouping of COAD patients according to gender: male (N = 153, 53.31%), female (N = 134, 46.69%). (C) Grouping of COAD patients according to race: black or African American (N = 57, 21.19%), white (N = 201, 74.72%).
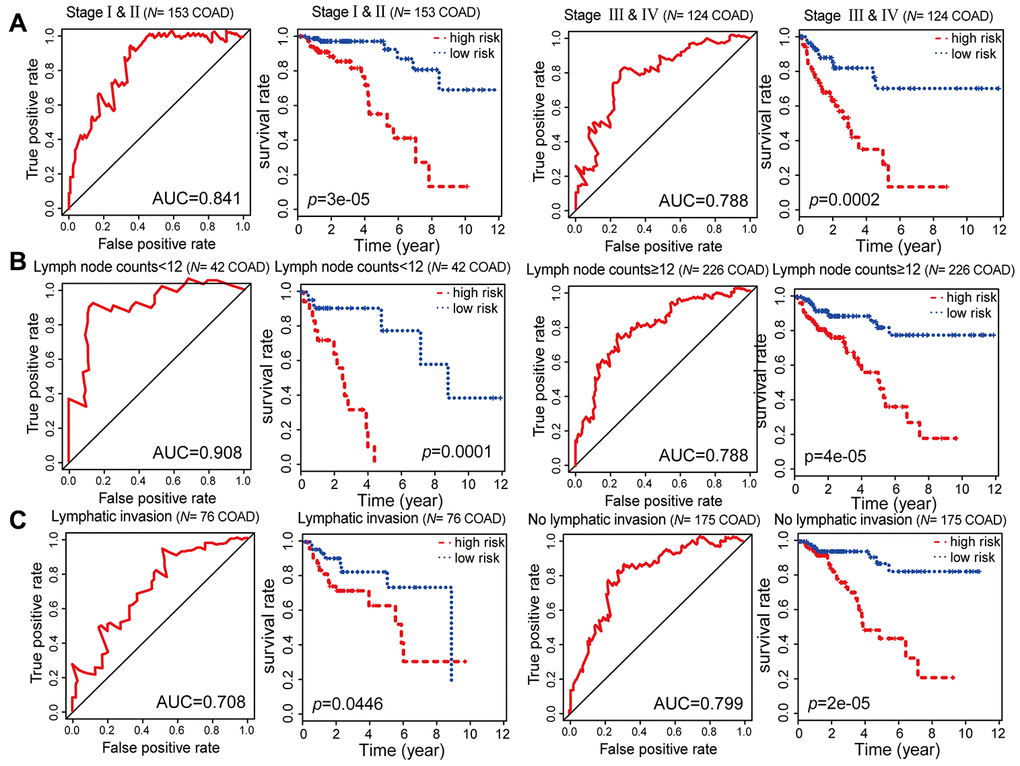
Figure 7. Kaplan-Meier and ROC analysis results based on stage, examined lymph node count and lymphatic invasion. (A) Grouping of COAD patients according to stage: early (stage I and II [N = 153, 53.31%]) and advanced (stage III and IV [N = 124, 43.21%]). (B) Grouping of COAD patients according to examined lymph node count: < 12 (N = 42, 14.63%) and ≥ 12 (N = 226, 78.75%). (C) Grouping of COAD patients according to lymphatic invasion: lymphatic invasion (N = 76, 26.48%) and no lymphatic invasion (N = 175, 60.98%).
In recent years, DNA methylation biomarkers have been increasingly recognized as important prognostic predictors in COAD. Previously identified markers of COAD prognosis have included hypermethylation of FAM134B [27], higher expression of MMP-11 [28], abnormal methylation and expression of DIRAS1 [29], upregulation of the long non-coding RNA BLACAT1 (a cell cycle regulator) [30] and abnormal expression of 10 microRNAs (including hsa-mir-891a, hsa-mir-6854, etc.) [31]. Dai et al. [32] demonstrated that combined biomarkers of DNA methylation were more sensitive and specific than individual DNA methylation markers. To compare the survival prediction ability of our combined prognostic predictive model with those of previously reported biomarkers, we performed ROC analyses of the previous mRNA, long non-coding RNA and microRNA biomarkers in the validation cohort. Our combined COAD prognostic model based on six CpG sites had a much higher AUC value than the other prognostic biomarkers assayed by ROC analysis in the COAD validation cohort from TCGA (N = 144) (Figure 8A). These results suggested that our combined COAD prognostic model provided more reliable and sensitive predictions of OS than other biomarkers in COAD patients.
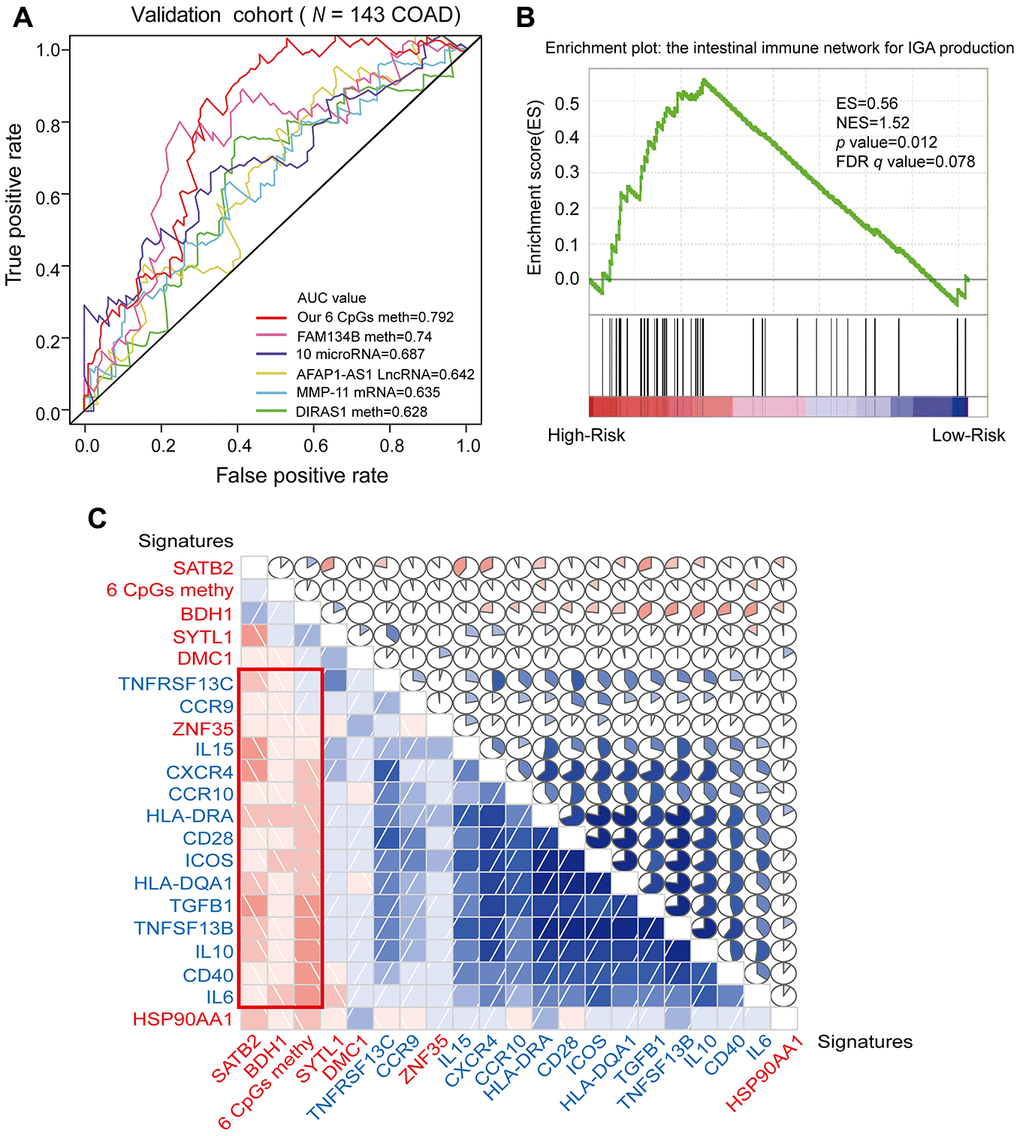
Figure 8. ROC analysis of different prognostic biomarkers and functional enrichment analysis of the corresponding genes. (A) ROC curve showing the sensitivity and specificity of our prognostic model and other known models in predicting the OS of patients in the validation cohort from TCGA. (B) COAD samples were divided into high-risk and low-risk groups, and the enrichment of IINIP pathway gene expression was analyzed using GSEA. ES, concentration fraction; NES, standardized ES; p-value, normalized p-value; FDR q-value, p-value corrected by the FDR method. (C) Correlation of the expression of the core enrichment genes from the IINIP pathway, the combined methylation level of our prognostic model and the expression of the genes corresponding to the individual CpG sites of the COAD prognostic biomarkers. The red signature represents the expression of the genes corresponding to the six CpG sites and the six-site combined methylation value; the blue signature represents the expression of the core enrichment genes in the IINIP pathway. Lower triangle: grids showing the correlation between two signatures, where blue indicates a positive correlation and red indicates a negative correlation. Upper triangle: circles represent the one-to-one correlation coefficients, differentiated by the fill area and intensity of shading. Blue indicates a positive correlation and red indicates a negative correlation.
Next, we performed a gene set enrichment analysis (GSEA) on the high- and low-risk groups that had been classified according to the median risk score. The pathways that correlated significantly with risk are illustrated in Figure 8B, Supplementary Figure 3 and Supplementary Table 7 (enrichment score [ES] > 0.4, |normalized enrichment score [NES]| > 1, p-value < 0.05 and FDR q-value < 0.25). We selected the intestinal immune network for IgA production (IINIP) for further analysis, since this pathway is known to be involved in COAD. The core enrichment genes of the IINIP pathway were obtained via GSEA (Supplementary Table 8). To determine whether the genes corresponding to the combined COAD prognostic biomarkers functioned in IINIP pathways, we conducted a one-to-one correlation analysis on the expression of the core enrichment genes from the IINIP pathway, the combined methylation level of our prognostic model and the expression of the genes corresponding to the individual CpG sites of the COAD prognostic biomarkers (Figure 8C and Supplementary Figure 4). As expected, the expression of BDH1 and SATB2 and the combined methylation level of our COAD prognostic model based on six CpG sites correlated significantly with the IINIP signaling pathway (p < 0.05). The above results indicated that our combined COAD prognostic model not only accurately predicted prognosis, but also included CpG sites that may directly or indirectly influence the IINIP pathway.
Discussion
This study was based on a comprehensive Illumina Infinium Human Methylation 450K array dataset in TCGA. To screen for potential diagnostic biomarkers, we first used 25 paired COAD and normal samples from TCGA to obtain Hyper-/Hypo-DMPs. Then, using conditional screening and machine learning based on these genome-wide DMPs, we obtained nine Hyper-DMPs and one Hypo-DMP as the final diagnostic biomarkers for inclusion in our COAD-specific diagnostic model. Our model could accurately and precisely distinguish COAD from normal tissues and nine types of cancerous tissues. Next, to screen for potential prognostic biomarkers, we performed a univariate Cox hazard regression analysis and a multivariate Cox stepwise regression analysis based on genome-wide CpG sites. We identified six CpG sites as potential prognostic biomarkers and used them to construct a combined COAD prognostic model. This model could predict the prognosis of COAD patients independently of important clinicopathological characteristics such as age, gender, race, AJCC stage, examined lymph node count and lymphatic invasion.
Combined DNA methylation models for the diagnosis of COAD have been constructed previously [24–26], and we compared our model with these models. A common problem with most of the previous COAD methylation diagnostic models was that they were only screened and constructed using COAD datasets. Although our diagnostic model was based on data from multiple cancer types, we could not identify DMPs that differentiated COAD from rectal cancer by this modeling method. Colon and rectal tumors were previously considered to differ in their epidemiology and treatment requirements [33]; however, newly published data from TCGA project [4] suggest that the overall patterns of methylation, mRNA and microRNA changes in colon and rectal cancers are indistinguishable. Thus, in future studies, we can try other modeling methods to distinguish COAD from rectal cancer and other cancers of the intestinal system.
We conducted in-depth KEGG and GO pathway enrichment analyses of the genes corresponding to the nine Hyper-DMPs and one Hypo-DMP, and we analyzed their protein-protein interactions in the STRING database. The leukemia inhibitory factor signaling pathway and the ciliary neurotrophic factor-mediated signaling pathway, both of which can activate the JAK2/STAT3 signaling pathway [34, 35], were found to be enriched in our GO analysis. Coincidentally, the JAK/STAT pathway was found to be enriched in our KEGG pathway analysis. JAK/STAT signaling, especially the overactivation of STAT3 and STAT5, is known to promote tumor cell survival, proliferation, and invasion [36]. Therefore, it is significant that our COAD-specific diagnostic biomarkers were both directly and indirectly associated with the JAK/STAT pathway.
The parameters of age, gender, race, AJCC stage, examined lymph node count and lymphatic invasion have been identified as important clinicopathological features of COAD prognosis. Specifically, age was found to be the most important prognostic factor for stage II COAD patients [13], women had a better prognosis than men [14], whites had a higher colorectal cancer survival rate than blacks [15], the AJCC TNM staging system was found to be a necessary adjuvant chemotherapy guide for stage II and III COAD patients [16], the examined lymph node count had excellent prognostic value for COAD patients undergoing surgery [17] and lymphatic invasion diagnosis was found to be an important indicator of lymph node metastasis in T1 COAD [18]. Since early-stage patients are more likely to be cured, prognostic markers that can effectively predict the risk of these patients will have higher clinical value [37]. Importantly, our combined prognostic prediction model based on six CpG sites was independent of these important clinicopathological characteristics of COAD, and had the potential to accurately predict the biological behavior of COAD at an early stage.
The six CpG sites included in our prognostic model were all CpG islands (dense clusters of CpG sites). Abnormal methylation of CpG islands is associated with the silencing of tumor suppressor genes. Two mechanisms have been proposed to explain the transcriptional inhibition caused by CpG island methylation. One proposed mechanism is that CpG islands directly impede the binding of specific transcription factors to recognition sites in promoters [38, 39]. The other proposed mechanism is that proteins that recognize methylated CpG sites, namely methyl CpG binding proteins, stimulate the inhibitory potential of methylated DNA [40].
When we searched the literature for information about the genes corresponding to the six CpG sites in our prognostic model (BDH1, SYTL1, SATB2, WDR20, DMC1 and ZNF35), we found that ZNF35 and SATB2 have already been established as reliable prognostic marker genes for COAD. For example, ZNF35 was found to differentiate the prognoses of COAD patients in a validation on independent test sets [41], and the transcription factor SATB2 was identified as a highly specific marker in colorectal adenocarcinoma when used in conjunction with CK20 [42]. On the other hand, low expression of DMC1 has been reported as a poor prognostic marker of ovarian cancer, together with high expression of XPC and RECQL [43]. These studies indirectly illustrate the reliability of our prognostic model.
Notably, GSEA revealed that our combined COAD prognostic model based on six CpG sites was significantly associated with core enrichment genes of the IINIP pathway, including HLA-DQB1, interleukin (IL)-6, IL-15 and CCR9. The IINIP pathway has been reported to alter the proliferation of COAD cells, the prognosis of COAD patients, the susceptibility of individuals to COAD, the effectiveness of immunotherapy for COAD, etc. [44–47]. These results suggested that our combined prognostic model could not only predict the prognosis of patients with COAD, but also reflect the immune pathways of COAD. Interestingly, IL-6 has been reported to participate with JAK2/STAT3 in a signaling pathway that promotes COAD cell proliferation [48], and the genes corresponding to the nine Hyper-DMPs and one Hypo-DMP in our COAD-specific diagnostic model were associated with the JAK/STAT pathway.
In summary, by analyzing the genome-wide methylation data of 299 COAD samples and 38 normal samples from TCGA, we found nine Hyper-DMPs and one Hypo-DMP that could be used as potential methylation biomarkers for the diagnosis of COAD. Our COAD-specific diagnostic model based on these DMPs not only differentiated COAD tissues from normal tissues with excellent accuracy and stability, but also precisely distinguished COAD from nine other cancer types (BLCA, BRCA, CESC, GBM, HNSC, LIHC, LUAD, LUSC and UCEC). Furthermore, using 287 COAD samples with prognostic information, we constructed a combined COAD prognostic evaluation model based on six CpG sites. Our model predicted the prognosis of COAD independently of important clinicopathological characteristics such as age, gender, race, AJCC stage, examined lymph node count and lymphatic invasion. Thus, both our COAD-specific diagnostic model and our combined prognostic model have high predictive capabilities and can be applied to the design of adjuvant chemotherapy clinical trials for early COAD patients.
Materials and Methods
Data source
We downloaded DNA methylation, gene expression and COAD clinical data from TCGA using the University of California Santa Cruz Xena tool (http://xena.ucsc.edu). The DNA methylation data were generated using the Illumina Human Methylation 450 Bead Chip platform, with methylation levels ranging from 0 to 1. We collected the methylation levels of ten types of tumor tissues and normal tissues from TCGA: COAD (299 tumors, 38 normal), BLCA (413 tumors, 21 normal), BRCA (790 tumors, 98 normal), CESC (309 tumors, 3 normal), UCEC (432 tumors, 46 normal), GBM (153 tumors, 2 normal), HNSC (530 tumors, 50 normal), LIHC (379 tumors, 50 normal), LUAD (460 tumors, 32 normal) and LUSC (372 tumors, 43 normal). We calculated the average methylation level of multiple samples as the methylation level of a given CpG site. The level-3 gene expression data were downloaded from RNA-seq HiSeqV2 based on the Illumina HiSeq 2000 RNA sequencing platform, and were obtained as RNA-Seq by Expectation-Maximization (RSEM) [49] normalized counts converted by log2 (x + 1). RSEM software was used to normalize counts on the Xena website.
In addition, we downloaded five DNA methylation array cohorts from GEO (https://www.ncbi.nlm.nih.gov/geo/) as independent validation cohorts. (1) The GSE42752 cohort [26] included 22 COAD samples and 41 normal samples from Russia. Quality control was performed using the R package GenomeStudio (v. 2011.1). Samples were removed if p was > 0.05 and the CpG coverage was < 95%. Thereafter, GenomeStudio was used to normalize the DNA methylation data. The methylation levels of the CpG sites were calculated as β values, where β = intensity (methylated)/intensity (methylated + unmethylated). The data were further normalized using a peak correction algorithm embedded in the IMA R package. Finally, CpG sites on sex chromosomes were removed, and the remaining CpG sites were retained for further analysis. (2) The GSE53051 cohort [50] included 35 COAD samples and 18 normal samples from the US. The R package minfi was used to preprocess the methylation data. To analyze the methylation levels of CpG sites, we averaged the values of all individuals in normal colon and colon cancer. We compared the data from colon cancer and normal control tissues using t-tests. We defined CpG sites with q values < 0.05 and a difference magnitude > 0.1 as important CpG sites. (3) The GSE77718 cohort [51] included 96 COAD samples and 96 normal samples from New Zealand. The CpG sites of each sample were preprocessed and rescaled to ensure that the internal control CpG sites had a common mean across samples. CpG sites located on sex chromosomes or known to cross-react with other regions of the genome were excluded from further analyses. The methylation of the remaining CpG sites was corrected using the COMBAT algorithm to account for batch effects (between-array effects). (4) The GSE48684 cohort [52] included 64 COAD samples and 41 normal samples from the US. CpG sites were removed if p was > 0.05 in the Illumina Infinium DNA methylation data. Thereafter, the R package minfi was used for normalization, including Illumina background level correction, color adjustment and Subset-quantile Within Array Normalization. CpG sites beginning with “rs” on the array were excluded, along with non-CpG sites associated with the X chromosome. The COMBAT algorithm was used to evaluate and correct the batch processing effects of all array runs. (5) The GSE77954 cohort [53] included 20 COAD samples and 11 normal samples from the US. The microarray data were collected at Expression Analysis Inc. (Durham, NC, USA) and preprocessed using the R package methylamine. The array platform was the Human Methylation 450 Bead Chip (GPL13534). We normalized the data from each cohort using the R package limma.
Difference and correlation analysis of DNA methylation and corresponding gene expression
Paired samples are the most suitable for assessing differential methylation levels among individuals [54]. Therefore, we used 25 paired samples to obtain DMPs. CpG sites with > 10% missing values were excluded during the screening process. Missing values in the remaining CpG sites were replaced by the median of the cohort. Then, a paired t-test was used to obtain DMPs between COAD and normal tissues, and the FDR values were adjusted by the Bonferroni method. CpG sites on the sex chromosomes were removed. CpG sites with |Δβ| > 0.2 and FDR values < 0.05 were considered differentially methylated. The R package Pheatmap [55] was used for heat mapping and unsupervised cluster analysis.
When a CpG site corresponded to multiple genes, the optimal corresponding gene was obtained using the R package Champ [56]. We used TCGA data from 25 paired patients with both COAD and normal expression profiles for differential gene expression analysis. The R package limma was used to identify differentially expressed genes from the original data. Genes with a log2 |fold change| > 1 and FDR < 0.05 were considered differentially expressed. Pearson correlation coefficients were calculated to assess the association between DNA methylation and gene expression. Correlations were considered significant based on an |r (cor-value)| > 0.1 and FDR < 0.05. All FDR values were adjusted by the Bonferroni method.
Acquisition of candidate diagnostic biomarkers and construction of a diagnostic model
The 299 COAD and 38 normal samples from TCGA were randomly assigned to the training and validation cohorts at a ratio of 2:1. Five independent GEO cohorts (GSE42752, GSE53051, GSE77718, GSE48684 and GSE77954) were also used as validation cohorts. Firstly, 25 paired COAD and normal samples were compared to obtain DMPs. Secondly, TCGA data from 10 cancer types were used to further screen and narrow the range of candidate DMPs. Candidate Hyper-DMPs were required to have an average methylation level in COAD that was 0.2 units higher than the average methylation level in the normal samples and the nine other types of tumor and normal samples (BLCA, BRCA, CESC, GBM, HNSC, LIHC, LUAD, LUSC and UCEC). Candidate Hypo-DMPs were required to have an average methylation level in COAD that was 0.2 units lower than the average methylation level in the normal samples and the nine other types of tumor and normal samples. Thirdly, we evaluated whether the candidate Hyper-/Hypo-DMPs were significantly associated with the expression of their corresponding genes (|r| > 0.1 and FDR < 0.05), and whether the corresponding genes were differentially expressed genes (log2 |fold change| > 1 and FDR < 0.05). Then, the selected attributes function in the data mining tool WEKA [57] was used to obtain the final list of potential diagnostic biomarkers (nine Hypo-DMPs and one Hypo-DMP). Lastly, for machine learning in WEKA, we used BayesNet to construct the COAD-specific diagnostic model with the nine Hyper-DMPs and one Hypo-DMP in the training cohort from TCGA (including 200 COAD and 25 normal samples).
Then, the diagnostic model was verified with the validation cohort from TCGA (including 99 COAD and 13 normal samples) and five independent GEO COAD cohorts. Firstly, we imported the methylation values of the nine Hyper-DMPs and one Hypo-DMP from the training cohort using the Filter option of the WEKA Preprocess panel. Then, we selected BayesNet in the Classifier option of the Classify panel to build our model. Thereafter, we imported the methylation values of the nine Hyper-DMPs and one Hypo-DMP from the validation cohort (TCGA validation set or GSE42752 or GSE53051 or GSE77718 or GSE48684 or GSE77954) to the Supplied test set option of the Classify panel. Finally, selecting BayesNet in the Classifier option, we identified the sensitivity, specificity, and accuracy of our model in the evaluated cohort.
Acquisition of candidate prognostic biomarkers and construction of a prognostic model
A total of 287 COAD tissue samples in the cohort from TCGA had both methylated β values and corresponding prognostic information. These samples were randomly assigned to the training cohort and the test cohort at a ratio of 1:1. Firstly, univariate Cox proportional hazard regression analysis was performed in the training cohort from TCGA (143 COAD samples) to identify significant methylation markers associated with the OS of COAD patients (p < 0.05). Then, multivariate Cox stepwise regression analysis was performed on these CpG sites, and sites with p-values > 0.05 were removed from the feature cohort in each iteration. The R packages Survival and Mass were jointly used to complete the multivariate Cox stepwise regression analysis.
The Cox proportional risk model was used to determine patients’ hazard ratios and corresponding 95% confidence intervals. The linear combination of model predictors weighted by their regression coefficients was used as the formula to predict the survival risk of patients. The high-risk and low-risk groups were classified according to the median risk value. The R packages Survival and Plot were then used to plot Kaplan-Meier survival curves to visualize the cumulative survival of the patients at risk at some time point. A log-rank test was used to evaluate the difference in OS between the high- and low-risk groups. Finally, the area under the ROC curve was determined by ROC analysis using the R package SurvivalROC. The effectiveness of the risk score in predicting OS was also evaluated. All statistical calculations were performed using the R statistical environment (R version 3.5.4).
STRING database
The genes corresponding to the diagnostic model were analyzed using the STRING functional protein-protein interaction network (9.1) [58]. The same website was used to analyze the input GO biological processes and KEGG pathways. P-values < 0.05 were considered significant.
GSEA
After the prognostic prediction model was used to calculate patients’ risk scores, GSEA (JAVA version) [59] (http://software.broadinstitute.org/gsea/index.jsp) was performed for the high-risk and low-risk groups. GSEA includes four key statistics: the ES, NES, FDR q-value and p-value. An ES > 0.4, |NES| > 1, p-value < 0.05 and FDR q-value < 0.25 were used to filter the GSEA results. Based on these statistics, all the genes in the list of specific genes were scored and ranked.
Supplementary Materials
Author Contributions
Y.W., H.W. and M.W. conceived and designed the project; H.W. and M.W. contributed analytic tools and/or grant support; Y.W., H.W., M.W., M.Z., X.H. and W.Q. prepared, wrote, reviewed and/or revised the manuscript. All authors discussed the results and commented on the manuscript.
Acknowledgments
The authors would like to acknowledge the Key Laboratory of Precision Diagnosis and Treatment of Gastrointestinal Tumors, Ministry of Education (China Medical University, Shenyang, China) for providing the space and equipment for this study.
Conflicts of Interest
The authors declare no competing financial interests.
Funding
This work was supported by grants from the National Natural Science Foundation of China (81872905, 31828005, 81673475), the National Natural Science Foundation of China and Liaoning Joint Fund Key Program (U1608281), the Liaoning Revitalization Talents Program (XLYC1807155), the China Postdoctoral Science Foundation (2019M661180 and 2019T120225), Shenyang S&T Projects (19-109-4-09), the Science and Technology Innovative Foundation for Young and Middle-aged Scientists of Shenyang City (RC200382) and the Key R&D Guidance Plan Projects in Liaoning Province (2019JH8/10300011).
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2019. CA Cancer J Clin. 2019; 69:7–34. https://doi.org/10.3322/caac.21551 [PubMed]
- 2. Rawla P, Sunkara T, Barsouk A. Epidemiology of colorectal cancer: incidence, mortality, survival, and risk factors. Prz Gastroenterol. 2019; 14:89–103. https://doi.org/10.5114/pg.2018.81072 [PubMed]
- 3. Ting WC, Chen LM, Pao JB, Yang YP, You BJ, Chang TY, Lan YH, Lee HZ, Bao BY. Common genetic variants in Wnt signaling pathway genes as potential prognostic biomarkers for colorectal cancer. PLoS One. 2013; 8:e56196. https://doi.org/10.1371/journal.pone.0056196 [PubMed]
- 4. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–37. https://doi.org/10.1038/nature11252 [PubMed]
- 5. Hong SN. Genetic and epigenetic alterations of colorectal cancer. Intest Res. 2018; 16:327–37. https://doi.org/10.5217/ir.2018.16.3.327 [PubMed]
- 6. Ewing I, Hurley JJ, Josephides E, Millar A. The molecular genetics of colorectal cancer. Frontline Gastroenterol. 2014; 5:26–30. https://doi.org/10.1136/flgastro-2013-100329 [PubMed]
- 7. Lao VV, Grady WM. Epigenetics and colorectal cancer. Nat Rev Gastroenterol Hepatol. 2011; 8:686–700. https://doi.org/10.1038/nrgastro.2011.173 [PubMed]
- 8. Paziewska A, Dabrowska M, Goryca K, Antoniewicz A, Dobruch J, Mikula M, Jarosz D, Zapala L, Borowka A, Ostrowski J. DNA methylation status is more reliable than gene expression at detecting cancer in prostate biopsy. Br J Cancer. 2014; 111:781–89. https://doi.org/10.1038/bjc.2014.337 [PubMed]
- 9. Gao D, Herman JG, Guo M. The clinical value of aberrant epigenetic changes of DNA damage repair genes in human cancer. Oncotarget. 2016; 7:37331–46. https://doi.org/10.18632/oncotarget.7949 [PubMed]
- 10. Ji H, Ehrlich LI, Seita J, Murakami P, Doi A, Lindau P, Lee H, Aryee MJ, Irizarry RA, Kim K, Rossi DJ, Inlay MA, Serwold T, et al. Comprehensive methylome map of lineage commitment from haematopoietic progenitors. Nature. 2010; 467:338–42. https://doi.org/10.1038/nature09367 [PubMed]
- 11. Sobhani I, Bergsten E, Couffin S, Amiot A, Nebbad B, Barau C, de’Angelis N, Rabot S, Canoui-Poitrine F, Mestivier D, Pédron T, Khazaie K, Sansonetti PJ. Colorectal cancer-associated microbiota contributes to oncogenic epigenetic signatures. Proc Natl Acad Sci USA. 2019; 116:24285–95. https://doi.org/10.1073/pnas.1912129116 [PubMed]
- 12. Beggs AD, Mehta S, Deeks JJ, James JD, Caldwell GM, Dilworth MP, Stockton JD, Blakeway D, Pestinger V, Vince A, Taniere P, Iqbal T, Magill L, et al, and ENDCAP-C Module 1 Collaborative. Validation of epigenetic markers to identify colitis associated cancer: results of module 1 of the ENDCAP-C study. EBioMedicine. 2019; 39:265–71. https://doi.org/10.1016/j.ebiom.2018.11.034 [PubMed]
- 13. Trivedi H, Chamarthy U, Dicarlo L, Herman J, Srkalovic G. Prognostic factors of overall survival for patients with stage II colon cancer. Acta Med Acad. 2014; 43:134–43. https://doi.org/10.5644/ama2006-124.112 [PubMed]
- 14. Seeland U, Heger U, Heise K, Ott K. [Gender aspects in gastrointestinal tumours and their prognosis in regard to multimodal treatment concepts]. Zentralbl Chir. 2015; 140:266–72. https://doi.org/10.1055/s-0035-1545963 [PubMed]
- 15. Andaya AA, Enewold L, Zahm SH, Shriver CD, Stojadinovic A, McGlynn KA, Zhu K. Race and colon cancer survival in an equal-access health care system. Cancer Epidemiol Biomarkers Prev. 2013; 22:1030–36. https://doi.org/10.1158/1055-9965.EPI-13-0143 [PubMed]
- 16. Liu Q, Luo D, Cai S, Li Q, Li X. Real-world implications of nonbiological factors with staging, prognosis and clinical management in colon cancer. Cancers (Basel). 2018; 10:263. https://doi.org/10.3390/cancers10080263 [PubMed]
- 17. Dolan RD, McSorley ST, Horgan PG, McMillan DC. Determinants of lymph node count and positivity in patients undergoing surgery for colon cancer. Medicine (Baltimore). 2018; 97:e0185. https://doi.org/10.1097/MD.0000000000010185 [PubMed]
- 18. Wada H, Shiozawa M, Sugano N, Morinaga S, Rino Y, Masuda M, Akaike M, Miyagi Y. Lymphatic invasion identified with D2-40 immunostaining as a risk factor of nodal metastasis in T1 colorectal cancer. Int J Clin Oncol. 2013; 18:1025–31. https://doi.org/10.1007/s10147-012-0490-9 [PubMed]
- 19. Lind GE, Guriby M, Ahlquist T, Hussain I, Jeanmougin M, Søreide K, Kørner H, Lothe RA, Nordgård O. Prognostic relevance of an epigenetic biomarker panel in sentinel lymph nodes from colon cancer patients. Clin Epigenetics. 2017; 9:97. https://doi.org/10.1186/s13148-017-0397-4 [PubMed]
- 20. Liang Y, Zhang C, Dai DQ. Identification of differentially expressed genes regulated by methylation in colon cancer based on bioinformatics analysis. World J Gastroenterol. 2019; 25:3392–407. https://doi.org/10.3748/wjg.v25.i26.3392 [PubMed]
- 21. Ahn JB, Chung WB, Maeda O, Shin SJ, Kim HS, Chung HC, Kim NK, Issa JP. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2011; 117:1847–54. https://doi.org/10.1002/cncr.25737 [PubMed]
- 22. Phipson B, Oshlack A. DiffVar: a new method for detecting differential variability with application to methylation in cancer and aging. Genome Biol. 2014; 15:465. https://doi.org/10.1186/s13059-014-0465-4 [PubMed]
- 23. Singh S, Singh R. Application of supervised machine learning algorithms for the classification of regulatory RNA riboswitches. Brief Funct Genomics. 2017; 16:99–105. https://doi.org/10.1093/bfgp/elw005 [PubMed]
- 24. Azuara D, Aussó S, Rodriguez-Moranta F, Guardiola J, Sanjuan X, Lobaton T, Boadas J, Piqueras M, Monfort D, Guinó E, Moreno V, Capellá G, de Oca J. New methylation biomarker panel for early diagnosis of dysplasia or cancer in high-risk inflammatory bowel disease patients. Inflamm Bowel Dis. 2018; 24:2555–64. https://doi.org/10.1093/ibd/izy255 [PubMed]
- 25. Beggs AD, James J, Caldwell G, Prout T, Dilworth MP, Taniere P, Iqbal T, Morton DG, Matthews G. Discovery and validation of methylation biomarkers for ulcerative colitis associated neoplasia. Inflamm Bowel Dis. 2018; 24:1503–09. https://doi.org/10.1093/ibd/izy119 [PubMed]
- 26. Naumov VA, Generozov EV, Zaharjevskaya NB, Matushkina DS, Larin AK, Chernyshov SV, Alekseev MV, Shelygin YA, Govorun VM. Genome-scale analysis of DNA methylation in colorectal cancer using infinium HumanMethylation450 BeadChips. Epigenetics. 2013; 8:921–34. https://doi.org/10.4161/epi.25577 [PubMed]
- 27. Islam F, Gopalan V, Pillai S, Lu CT, Kasem K, Lam AK. Promoter hypermethylation inactivate tumor suppressor FAM134B and is associated with poor prognosis in colorectal cancer. Genes Chromosomes Cancer. 2018; 57:240–51. https://doi.org/10.1002/gcc.22525 [PubMed]
- 28. Pang L, Wang DW, Zhang N, Xu DH, Meng XW. Elevated serum levels of MMP-11 correlate with poor prognosis in colon cancer patients. Cancer Biomark. 2016; 16:599–607. https://doi.org/10.3233/CBM-160601 [PubMed]
- 29. Zheng R, Gao D, He T, Zhang M, Zhang X, Linghu E, Wei L, Guo M. Methylation of DIRAS1 promotes colorectal cancer progression and may serve as a marker for poor prognosis. Clin Epigenetics. 2017; 9:50. https://doi.org/10.1186/s13148-017-0348-0 [PubMed]
- 30. Su J, Zhang E, Han L, Yin D, Liu Z, He X, Zhang Y, Lin F, Lin Q, Mao P, Mao W, Shen D. Long noncoding RNA BLACAT1 indicates a poor prognosis of colorectal cancer and affects cell proliferation by epigenetically silencing of p15. Cell Death Dis. 2017; 8:e2665. https://doi.org/10.1038/cddis.2017.83 [PubMed]
- 31. Wei HT, Guo EN, Liao XW, Chen LS, Wang JL, Ni M, Liang C. Genome-scale analysis to identify potential prognostic microRNA biomarkers for predicting overall survival in patients with colon adenocarcinoma. Oncol Rep. 2018; 40:1947–58. https://doi.org/10.3892/or.2018.6607 [PubMed]
- 32. Dai W, Teodoridis JM, Zeller C, Graham J, Hersey J, Flanagan JM, Stronach E, Millan DW, Siddiqui N, Paul J, Brown R. Systematic CpG islands methylation profiling of genes in the Wnt pathway in epithelial ovarian cancer identifies biomarkers of progression-free survival. Clin Cancer Res. 2011; 17:4052–62. https://doi.org/10.1158/1078-0432.CCR-10-3021 [PubMed]
- 33. Minsky BD. Unique considerations in the patient with rectal cancer. Semin Oncol. 2011; 38:542–51. https://doi.org/10.1053/j.seminoncol.2011.05.008 [PubMed]
- 34. Seto DN, Kandarian SC, Jackman RW. A key role for leukemia inhibitory factor in C26 cancer cachexia. J Biol Chem. 2015; 290:19976–86. https://doi.org/10.1074/jbc.M115.638411 [PubMed]
- 35. Wang L, Zhang H, Lei D. microRNA-146a promotes growth of acute leukemia cells by downregulating ciliary neurotrophic factor receptor and activating JAK2/STAT3 signaling. Yonsei Med J. 2019; 60:924–34. https://doi.org/10.3349/ymj.2019.60.10.924 [PubMed]
- 36. Groner B, von Manstein V. Jak stat signaling and cancer: opportunities, benefits and side effects of targeted inhibition. Mol Cell Endocrinol. 2017; 451:1–14. https://doi.org/10.1016/j.mce.2017.05.033 [PubMed]
- 37. Segura MF, Belitskaya-Lévy I, Rose AE, Zakrzewski J, Gaziel A, Hanniford D, Darvishian F, Berman RS, Shapiro RL, Pavlick AC, Osman I, Hernando E. Melanoma MicroRNA signature predicts post-recurrence survival. Clin Cancer Res. 2010; 16:1577–86. https://doi.org/10.1158/1078-0432.CCR-09-2721 [PubMed]
- 38. Singal R, Ginder GD. DNA methylation. Blood. 1999; 93:4059–70. https://doi.org/10.1182/blood.V93.12.4059 [PubMed]
- 39. Tate PH, Bird AP. Effects of DNA methylation on DNA-binding proteins and gene expression. Curr Opin Genet Dev. 1993; 3:226–31. https://doi.org/10.1016/0959-437x(93)90027-m [PubMed]
- 40. Kass SU, Pruss D, Wolffe AP. How does DNA methylation repress transcription? Trends Genet. 1997; 13:444–49. https://doi.org/10.1016/s0168-9525(97)01268-7 [PubMed]
- 41. Yin Z, Yan X, Wang Q, Deng Z, Tang K, Cao Z, Qiu T. Detecting prognosis risk biomarkers for colon cancer through multi-omics-based prognostic analysis and target regulation simulation modeling. Front Genet. 2020; 11:524. https://doi.org/10.3389/fgene.2020.00524 [PubMed]
- 42. Ma C, Olevian D, Miller C, Herbst C, Jayachandran P, Kozak MM, Chang DT, Pai RK. SATB2 and CDX2 are prognostic biomarkers in DNA mismatch repair protein deficient colon cancer. Mod Pathol. 2019; 32:1217–31. https://doi.org/10.1038/s41379-019-0265-1 [PubMed]
- 43. Sun H, Cao D, Ma X, Yang J, Peng P, Yu M, Zhou H, Zhang Y, Li L, Huo X, Shen K. Identification of a prognostic signature associated with DNA repair genes in ovarian cancer. Front Genet. 2019; 10:839. https://doi.org/10.3389/fgene.2019.00839 [PubMed]
- 44. Wang K, Yu X, Jiang H, Huang J, Wang H, Jiang H, Wei S, Liu L. Genome-wide expression profiling-based copy number variations and colorectal cancer risk in Chinese. Mol Carcinog. 2019; 58:1324–33. https://doi.org/10.1002/mc.23015 [PubMed]
- 45. Chang YC, Su CY, Chen MH, Chen WS, Chen CL, Hsiao M. Secretory RAB GTPase 3C modulates IL6-STAT3 pathway to promote colon cancer metastasis and is associated with poor prognosis. Mol Cancer. 2017; 16:135. https://doi.org/10.1186/s12943-017-0687-7 [PubMed]
- 46. Bondurant KL, Lundgreen A, Herrick JS, Kadlubar S, Wolff RK, Slattery ML. Interleukin genes and associations with colon and rectal cancer risk and overall survival. Int J Cancer. 2013; 132:905–15. https://doi.org/10.1002/ijc.27660 [PubMed]
- 47. Löfroos AB, Kadivar M, Resic Lindehammer S, Marsal J. Colorectal cancer-infiltrating T lymphocytes display a distinct chemokine receptor expression profile. Eur J Med Res. 2017; 22:40. https://doi.org/10.1186/s40001-017-0283-8 [PubMed]
- 48. Fang X, Hong Y, Dai L, Qian Y, Zhu C, Wu B, Li S. CRH promotes human colon cancer cell proliferation via IL-6/JAK2/STAT3 signaling pathway and VEGF-induced tumor angiogenesis. Mol Carcinog. 2017; 56:2434–45. https://doi.org/10.1002/mc.22691 [PubMed]
- 49. Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011; 12:323. https://doi.org/10.1186/1471-2105-12-323 [PubMed]
- 50. Timp W, Bravo HC, McDonald OG, Goggins M, Umbricht C, Zeiger M, Feinberg AP, Irizarry RA. Large hypomethylated blocks as a universal defining epigenetic alteration in human solid tumors. Genome Med. 2014; 6:61. https://doi.org/10.1186/s13073-014-0061-y [PubMed]
- 51. McInnes T, Zou D, Rao DS, Munro FM, Phillips VL, McCall JL, Black MA, Reeve AE, Guilford PJ. Genome-wide methylation analysis identifies a core set of hypermethylated genes in CIMP-H colorectal cancer. BMC Cancer. 2017; 17:228. https://doi.org/10.1186/s12885-017-3226-4 [PubMed]
- 52. Luo Y, Wong CJ, Kaz AM, Dzieciatkowski S, Carter KT, Morris SM, Wang J, Willis JE, Makar KW, Ulrich CM, Lutterbaugh JD, Shrubsole MJ, Zheng W, et al. Differences in DNA methylation signatures reveal multiple pathways of progression from adenoma to colorectal cancer. Gastroenterology. 2014; 147:418–29.e8. https://doi.org/10.1053/j.gastro.2014.04.039 [PubMed]
- 53. Qu X, Sandmann T, Frierson H
Jr , Fu L, Fuentes E, Walter K, Okrah K, Rumpel C, Moskaluk C, Lu S, Wang Y, Bourgon R, Penuel E, et al. Integrated genomic analysis of colorectal cancer progression reveals activation of EGFR through demethylation of the EREG promoter. Oncogene. 2016; 35:6403–15. https://doi.org/10.1038/onc.2016.170 [PubMed] - 54. Stevens JR, Herrick JS, Wolff RK, Slattery ML. Power in pairs: assessing the statistical value of paired samples in tests for differential expression. BMC Genomics. 2018; 19:953. https://doi.org/10.1186/s12864-018-5236-2 [PubMed]
- 55. Dailey AL. Metabolomic bioinformatic analysis. Methods Mol Biol. 2017; 1606:341–52. https://doi.org/10.1007/978-1-4939-6990-6_22 [PubMed]
- 56. Tian Y, Morris TJ, Webster AP, Yang Z, Beck S, Feber A, Teschendorff AE. ChAMP: updated methylation analysis pipeline for illumina BeadChips. Bioinformatics. 2017; 33:3982–84. https://doi.org/10.1093/bioinformatics/btx513 [PubMed]
- 57. Smith TC, Frank E. Introducing machine learning concepts with WEKA. Methods Mol Biol. 2016; 1418:353–78. https://doi.org/10.1007/978-1-4939-3578-9_17 [PubMed]
- 58. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013; 41:D808–15. https://doi.org/10.1093/nar/gks1094 [PubMed]
- 59. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for gene set enrichment analysis. Bioinformatics. 2007; 23:3251–53. https://doi.org/10.1093/bioinformatics/btm369 [PubMed]