




Introduction
Senescent cells lose their proliferative potential in response to various stresses. They secrete a variety of pro-inflammatory mediators and proteases, gathered in the senescence-associated secretory phenotype (SASP) [1] that engages the immune system to eliminate senescent cells [2,3]. Senescent cells accumulate in aging organisms, chronic age-related diseases and benign tumors [4–7]; conversely, elimination of senescent cells contributes to improve health [8–11]. They also accumulate in tissues affected by atherosclerosis [12–14] and their elimination strikingly reduces atherogenicity in animal models [12,14]. Senescence is thus a link between molecular damage and the altered physiology of aging, and targeting SnC using senolytic drugs appears a promising strategy to reduce the burden of age-related chronic inflammatory diseases [15], including atherosclerosis [16,17]. Selective and safe senolytics, however, have yet to be discovered as most have emerged from the oncology therapeutic armamentarium [18]. Combination of the natural product quercetin with the tyrosine kinase inhibitor dasatinib preferentially killed senescent cells in culture and in mice, improving health span in naturally aged mice [19,20]. ABT263, a Bcl2 family inhibitor, was able to eliminate senescent cells after irradiation in mice and to rejuvenate bone marrow stem cells from both irradiated mice and naturally aged mice [21], to reverse pulmonary fibrosis in a mouse model [22], and to reduce senescence-associated Tau-dependent neuronal damage and cognitive decline [23].
Angiopoietin like-2 (angptl2) is a member of the SASP [24–28] and is detectable in most organs of adult mice [29]. Angptl2 is expressed by senescent vascular human endothelial cells (EC) [30], but not quiescent or proliferative EC [31] and is atherogenic when infused in young LDLr-/-;hApoB100-/- atherosclerotic (ATX) mice [31] or in angptl2-endothelial transgenic ApoE-/- mice [32]. We reported that plasma levels of angptl2 are elevated in patients with cardiovascular diseases (CVD) [31], were associated with endothelial dysfunction [33] and were predictive of major cardiac adverse events (MACE) and death [34]. Recently, we reported a strong relationship between arterial expression of p21, a cell cycle inhibitor overexpressed in senescent cells and maintaining growth arrest [35], and circulating levels of angptl2 in atherosclerotic patients [36]. Senescent EC are activated and promote aggregation of leukocytes [37], the initiating step of atherogenesis [38]. We therefore hypothesized that down-regulation of vascular angptl2, preferentially in the endothelium of severely dyslipidemic ATX mice would promote endothelial repair and slow atherogenesis. Here, we report that knockdown of vascular angptl2 by a shRNA (shAngptl2), delivered to the vascular cells via a single injection of an AAV1 [39], slowed atheroma progression in ATX mice. Knockdown of angptl2 was associated with a rapid reduction in the expression of EC senescence-associated p21 accompanied by the increase in Bax/Bcl2 ratio as a marker of apoptosis; subsequently, this was associated with endothelial repair as evidenced by the incorporation of endothelial progenitor CD34+ cells. In addition to our pre-clinical results, we show that vascular ANGPTL2 gene expression is correlated with p21 expression and inflammatory cytokines in the internal mammary artery isolated from severely atherosclerotic patients undergoing a coronary artery bypass surgery. Altogether, our data suggest that targeting vascular angptl2 could be senolytic, delaying the progression of atherosclerosis.
Results
Endothelial expression of angptl2 and senescence gene markers parallels atherogenesis
Firstly, and as expected, endothelial expression of angptl2, Pai-1 and p21 parallels the growing atheroma plaque in untreated LDLr-/-;hApoB100+/+ atherosclerotic (ATX) mice up to 12-month old (-mo) (Figure 1); p21 is a cyclin-dependent kinase inhibitor overexpressed in growth-arrested senescent cells, and Pai-1 is a recognized SASP member and inducer of senescence [15]. When compared to age-matched wild-type mice, angptl2, Pai-1 and p21 are over-expressed in the native endothelium of 6-mo ATX mice (Figure 2).
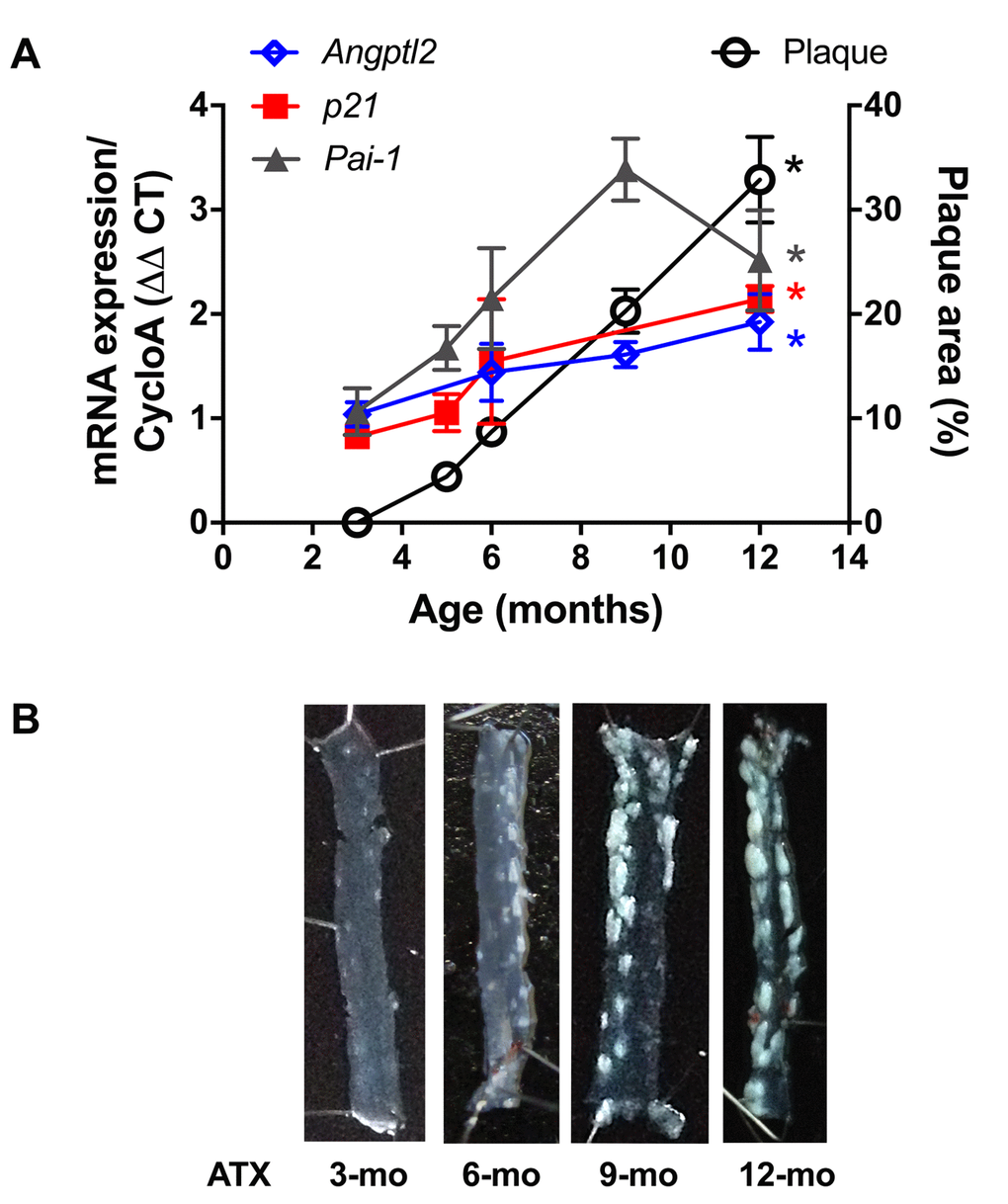
Figure 1. Age-dependent increase of senescence-associated p21, Pai-1 and angptl2 expressions in the native endothelium parallels plaque growth in the aorta. (A) mRNA expression of indicated genes was quantified in the native aortic endothelium of 3-mo (n=4), 5-mo (n=4), 6-mo (n=4) and 12-mo (n=4) control ATX mice. The average level of gene expression in 3-mo ATX mice was arbitrarily set at 1. Plaque area was quantified from longitudinally open thoracic aortas of 3-mo (n=7), 5-mo (n=5), 6-mo (n=7), 9-mo (n=12) and 12-mo (n=4) ATX mice. Data are expressed as mean±SEM. *: p<0.0001 vs. 3-mo ATX mice. (B) Representative pictures of age-related increase in atherosclerotic plaque in 3-, 6-, 9- and 12-mo ATX mice.
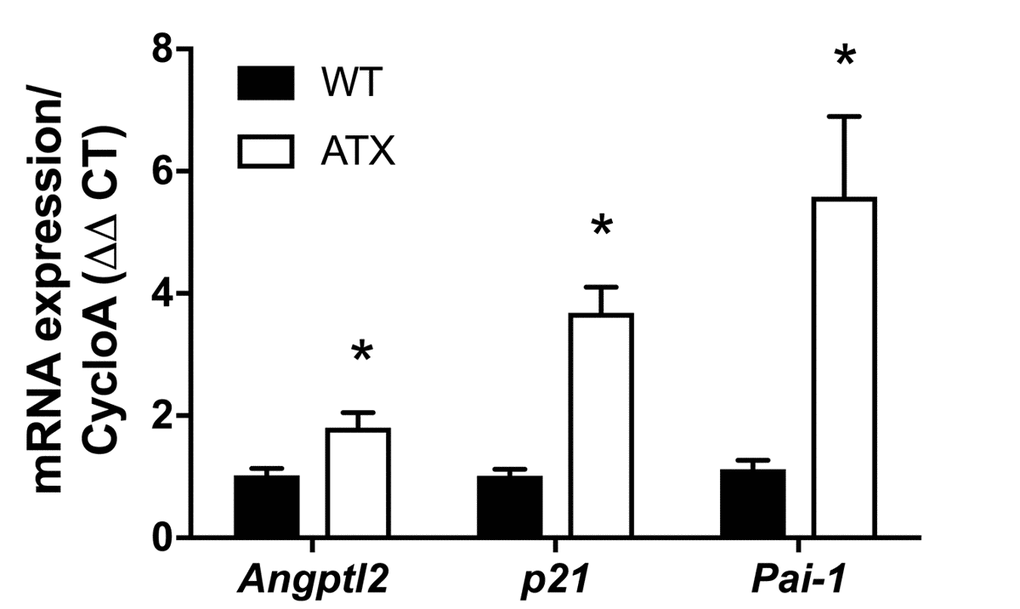
Figure 2. Increased expression of senescence-associated p21, Pai-1 and angptl2 in the native aortic endothelium of 6-month old ATX compared to WT mice. mRNA expression of indicated genes was quantified in the native aortic endothelium of 6-mo WT and ATX mice (n=3). The average level of gene expression in 6-mo WT mice was arbitrarily set at 1. Data are expressed as mean±SEM. *: p<0.05 vs. WT mice.
Vascular angptl2 knockdown decreases atherosclerotic plaque size
To investigate the anti-atherogenic effects of angptl2 knockdown, we delivered once a shAngptl2 (Table S1) using an adeno-associated virus serotype 1 (AAV1) as a vector (i.v. bolus injection) with preferred vascular tropism [39] in 3-mo ATX mice. Each mouse was sacrificed at 6-mo. The vascular delivery of the shRNA was confirmed by mCherry staining of the aortic wall, showing red fluorescence in the endothelial cells and throughout the vascular wall, but with no diffusion to the adventitia or in the plaque (Figure 3A). In addition, the AAV1-shAngptl2 infection neither reduced angptl2 expression in the mouse heart and liver (Figure 3B), nor affected lipid and glucose blood levels (Figure 3C).
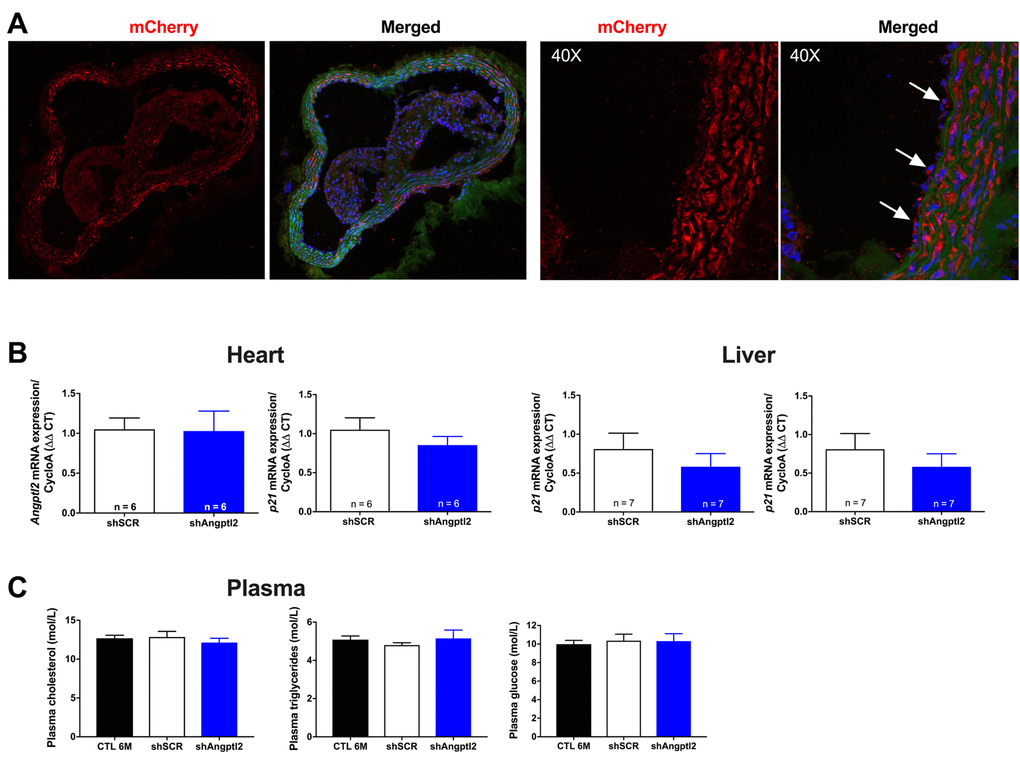
Figure 3. Distribution of the AAV1-mCherry in the aortic wall and specificity of the AAV1-shAngptl2. (A) Immunofluorescence of AAV1-mCherry in frozen aortic sections of ATX mice at 6 months of age, 3 months post-infection: mCherry signal distributed throughout the vascular wall is shown in red and basal lamina in green; nuclei are shown in blue. At a higher magnification (40X), arrows show mCherry signal in the endothelium. A negative control (absence of primary antibody against mCherry) was performed (data not shown). (B) Neither cardiac nor liver Angptl2 and p21 mRNA expressions were affected by the AAV1-shAngptl2 in ATX mice, 3 months post-infection. Average gene expression level in shSCR mice was arbitrarily set at 1. Data are mean±SEM of n ATX mice. C) Cholesterol, triglycerides and glucose levels of ATX mice were not altered by the AAV1-shAngptl2, 3 months post-infection. Data are mean±SEM of n=7 ATX mice.
Plaque was not present at 3-mo (Figures 1B and 4A), but the atherosclerotic lesion covered 8±1% of the thoracic aorta of 6-mo untreated ATX mice, and 8±1% of the thoracic aorta of mice injected with an AAV1 containing a scrambled (SCR; Table S1) mRNA sequence (Figure 4). At 6-mo, 3 months AAV1-shAngptl2 post-injection, the atherosclerotic burden in the thoracic aorta was reduced by 58% (Figure 4; p<0.0001; n=12) compared to un-injected ATX mice and mice injected with an AAV1-shSCR. Therefore, targeting angptl2+ vascular cells is anti-atherogenic.
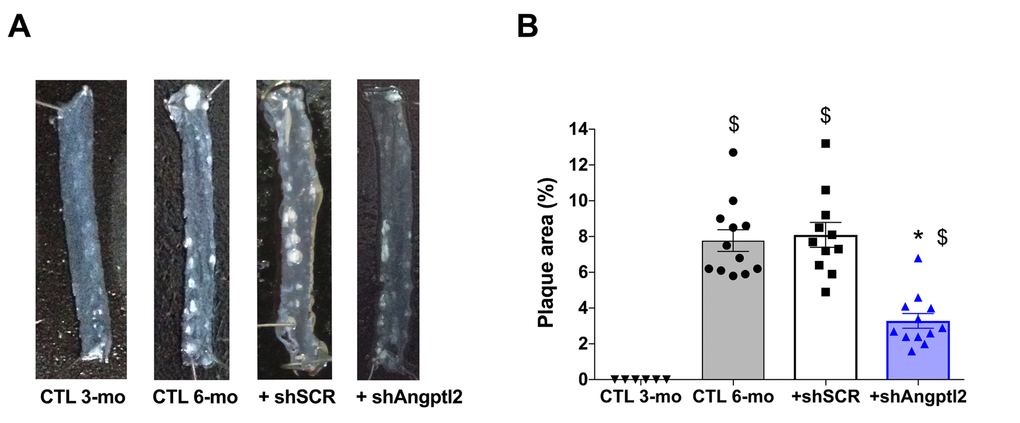
Figure 4. Down-regulation of vascular angptl2 is anti-atherogenic. (A) Representative images of longitudinally open thoracic aortas showing atherosclerotic plaque, and (B) Quantification of thoracic aorta atheroma plaque area in 3 mo control (CTL 3-mo, n=6), 6-mo control (CTL 6-mo, n=12), 6-mo AAV1-shScramble (+ shSCR, n=11) and 6-mo AAV1-shAngptl2 (+ shAngptl2, n=12) ATX mice revealing white atheroma plaques at 6-mo. Data are mean±SEM. *: p<0.0001 vs. CTL 6-mo and shSCR; $: p<0.0001 vs. CTL 3-mo.
Vascular Angptl2 down-regulation decreases markers of senescence, monocytes recruitment and inflammation in the endothelium
Some ATX mice were sacrificed 1, 2 and 4 weeks after AAV1-shAngptl2 infection. After only one week, endothelial expression of angptl2 and p21 were significantly lowered and continued to decline over the 4-week period post-AAV1-shAngptl2 injection (Figure 5), demonstrating a fast reduction in senescence. At the end of the treatment, 3 months post-AAV1-shAngptl2 injection, angptl2, p21 (Figures 5 and 6A) and Pai-1 expressions were still significantly decreased in the endothelium (Figure 6A), suggesting that knockdown of angptl2 lowers senescence in the long-term. Therefore, targeting angptl2+ EC reduces senescent EC load in vivo; conversely, this suggests that angptl2+ EC are senescent.
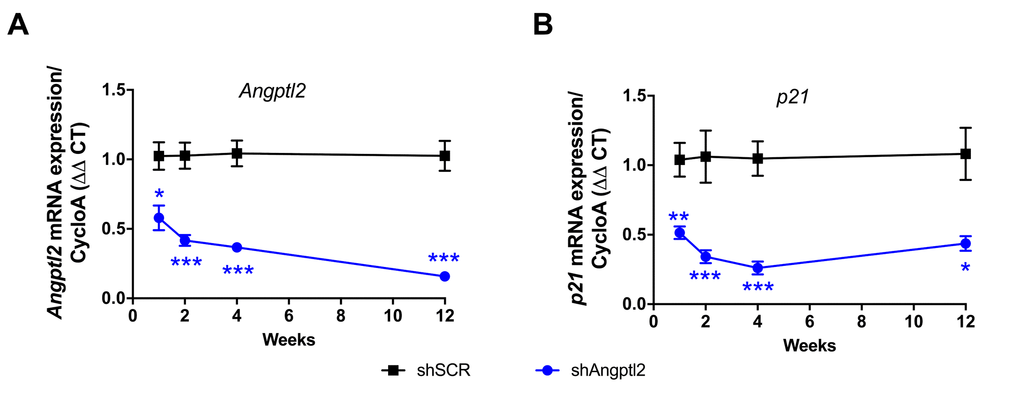
Figure 5. Time-dependent decrease of senescent-associated Angptl2 and p21 mRNA expressions inthe native endothelium of the aorta post-AAV1-shAngptl2 injection. Decrease of Angptl2 (A) and p21 (B) mRNA expression post-injections (1-week, n=5-6; 2-week, n=5-7; 4-week, n=4-7; 3-month, n=6-4) in the native endothelium freshly harvested from thoracic aortas of ATX mice treated with AAV1-shSCR or AAV1-shAngptl2. Average gene expression level in shSCR group was arbitrarily set at 1. Data are mean±SEM. *: p<0.05; **: p<0.01; ***: p<0.001 vs. shSCR group, at each time point.
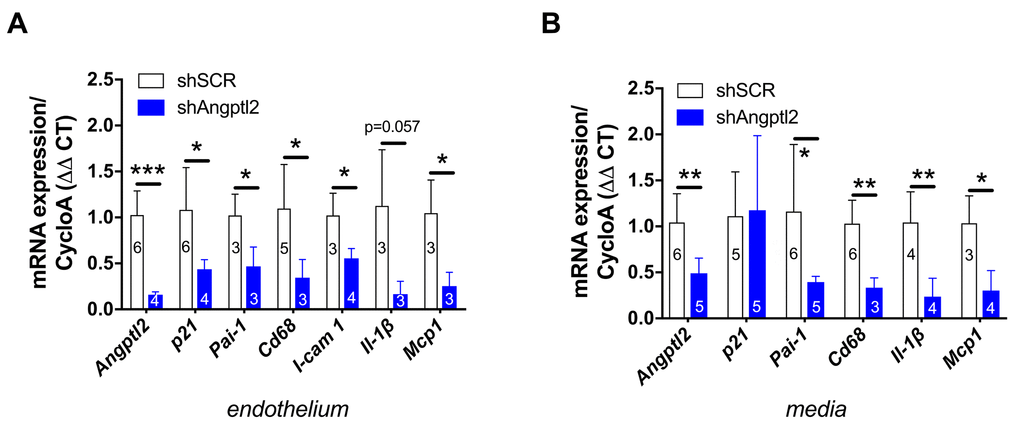
Figure 6. Down-regulation of vascular angptl2 is senolytic and anti-atherogenic. (A) Decrease of mRNA expression of the indicated genes in the native aortic endothelium isolated from AAV1-shAngptl2-treated ATX mice, 3 months post-infection. Data are mean±SEM of n mice. *: p<0.05; **: p<0.01; ***: p<0.001 vs. shSCR group. (B) Decrease of mRNA expression (except for p21) in the de-endothelialized aortic wall (media) isolated from AAV1-shAngptl2-treated ATX mice, 3 months post-infection. Data are mean±SEM. *: p<0.05 and **p<0.01 vs. shSCR group.
Senescent EC are known to favour adhesion of monocytes and their migration into the intima [37]. Accordingly, in the endothelium of 6-mo treated AAV1-shAngptl2 mice the expressions of the adhesion molecule Icam-1 and of monocyte chemo-attractant protein Mcp1 were reduced (Figure 6A) and this was associated with a lower expression of Cd68+ in endothelial cells (Figure 6A); the expression of the inflammatory marker IL-1β also tented (p=0.057) to decrease in AAV1-shAngptl2 mice (Figure 6A). Altogether, these data suggest that targeting angptl2+ senescent EC reduces inflammation and adhesion of Cd68+ monocytes onto the endothelium.
Vascular Angptl2 down-regulation decreases inflammation, monocytes recruitment and macrophages infiltration into the media of the aorta
In the present study, p21 expression was not altered in the media (including the atheroma plaque) of the aorta 3 months post-AAV1-shAngptl2 infection (Figure 6B), suggesting that in this mouse model and at this young age, arterial smooth muscle cells, fibroblasts and foam cells are not senescent. Angptl2 and Cd68 expression, however, decreased (Figure 6B), as did Pai-1, Il-1β and Mcp1, suggestive of less macrophage infiltration into the media and a lower inflammation. Indirectly, it also suggests that Cd68+ macrophages express angptl2 and contribute to the local inflammation, as previously reported [29,32,40]. Therefore, targeting angptl2+ vascular cells reduces inflammation associated with macrophages infiltration into the media.
Vascular Angptl2 down-regulation induces early apoptosis of senescent ECs and promotes endothelium repair
To determine the mechanism of senescent EC clearance and endothelial repair associated with the anti-atherogenic effect of the shAngptl2, we euthanized ATX mice 1, 2 and 4 weeks after AAV1-shAngptl2 infection. Concomitantly with the fast reduction in senescence (Figure 5), after only one week post-infection, endothelial pro-apoptotic Bax expression was significantly increased (p=0.005), while that of the anti-apoptotic Bcl2 was significantly decreased (p=0.003), increasing the Bax/Bcl2 ratio (Figure 7A), suggesting that angptl2+ senescent EC were eliminated, at least partly, following activation of this apoptotic pathway. In contrast, in the media neither Bax nor Bcl2 expression changed one week post-infection (Figure 7B), and this absence of apoptosis paralleled the lack of reduction in the senescence marker p21 in the media (Figure 6). Thus, AAV1-shAngptl2 infection promotes cell death of senescent angptl2+ endothelial cells by apoptosis; in contrast, in other cells from the aortic wall, shAngptl2 does not affect senescence and does not induce apoptosis.
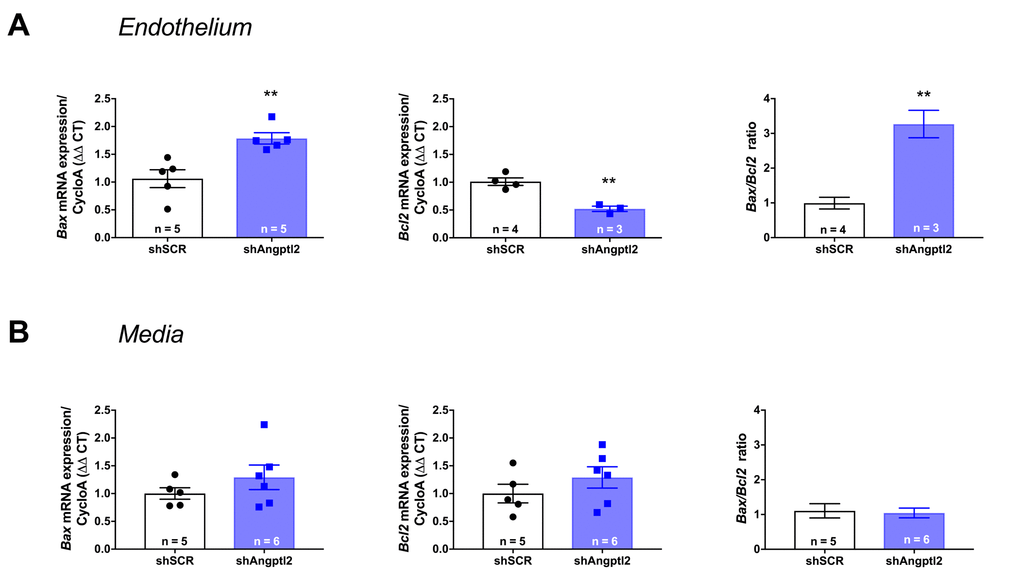
Figure 7. Down-regulation of vascular angptl2 is senolytic by promoting apopotosis. (A) Induction of Angptl2+ senescent endothelial cells apoptosis 1-week post-injection of AAV1-shAngptl2 in aortas of ATX mice: Bax mRNA expression increased (left, p=0.005) while Bcl2 mRNA expression decreased (middle, p=0.003), increasing Bax/Bcl2 ratio (right). (B) Absence of apoptosis in cells from the media 1-week post-injection of AAV1-shAngptl2 in aortas of ATX mice. Data are mean±SEM of n ATX mice.
Since the shAngptl2 therapy is anti-atherogenic, it would be expected that endothelial repair occurred as a mean to restore and/or maintain endothelial functions. After 2 weeks of shAngptl2 treatment, Cd34 expression tended to increase in the endothelium (from 1.1±0.3 to 1.9±0.3, p=0.082, n=5 and 7, respectively; data not shown), and after 4 weeks Cd34 expression increased significantly (p=0.034) (Figure 8A). This increase in the marker of progenitor cells, suggesting Cd34+ progenitor cell recruitment, was maintained up to the end of the study period, i.e. 3 months post-injection (Figure 8B-C). In contrast, endothelial apoptosis was no longer observed 3 months post-injection (data not shown). This strongly suggests that rapid apoptosis of angptl2+ senescent EC stimulates long-term endothelial repair, at least in part by incorporating circulating endothelial progenitor cells (CD34+ EpC).
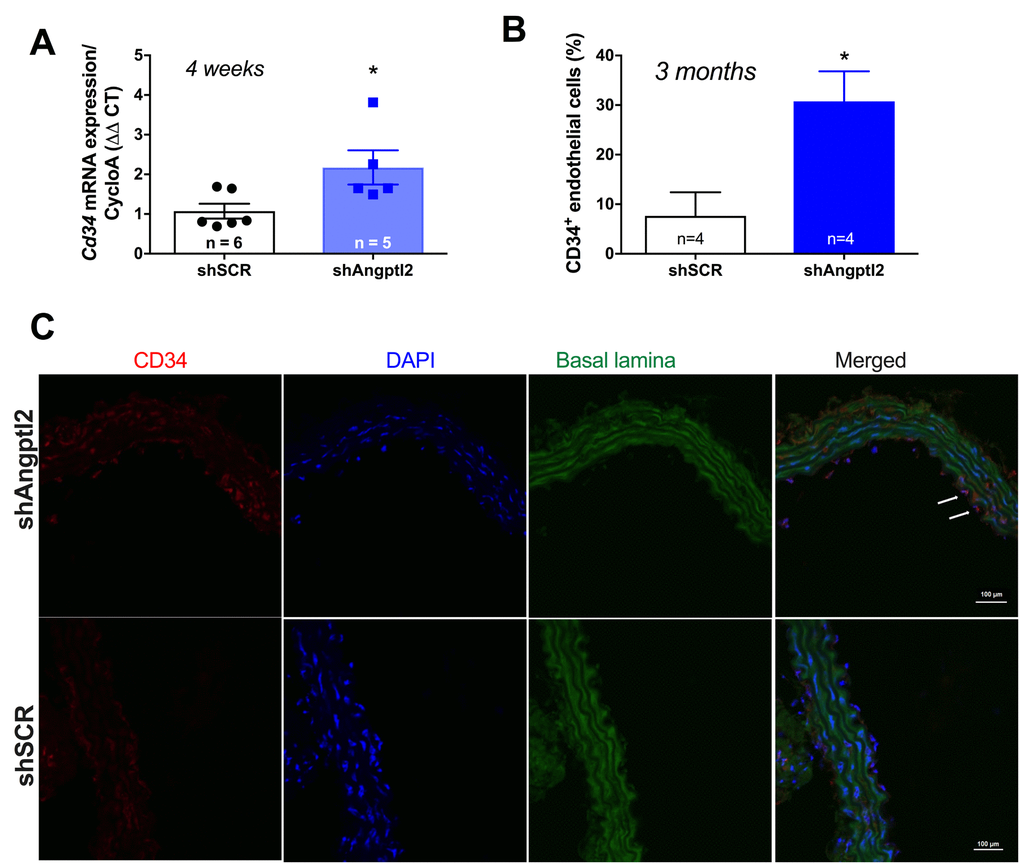
Figure 8. Down-regulation of vascular angptl2 is anti-atherogenic and promotes endothelial repair. (A) Increased expression of the progenitor marker Cd34 mRNA in the native aortic endothelium 4-week post-injection of shAngptl2 ATX mice. Data are mean±SEM. * : p<0.05 vs. shSCR group. The average gene expression levels in the shSCR group were arbitrarily set at 1. (B) Higher CD34 staining in the endothelium of 6-month old ATX mice 3 months post-injection of the AAV1-shAngptl2 compared to ATX mice injected with the scramble shRNA (shSCR). Data are mean±SEM. *: p<0.05 vs. shSCR. C) Representative images of CD34 immunostaining are shown.
Vascular ANGPTL2 expression correlates with vascular senescence in internal mammary artery of patients with severe coronary artery disease
To validate the clinical relevance of ANGPTL2 as a marker of vascular senescence, and to assess whether accumulation of senescent cells precedes plaque growth in human arteries, we measured the expression of ANGPTL2 and markers of senescence in human arterial segments. We recruited 26 patients undergoing coronary artery bypass (CABG) surgery with or without valve replacement (characteristics of the patients are in Table S2). In internal mammary artery segments free of plaque, the expression of p21 and that of ANGPTL2 were tightly associated (Figure 9A) and did not correlate with the age of the donor (data not shown). Furthermore, ANGPTL2 and p21 levels correlated with pro-inflammatory cytokines of the SASP, i.e., TNFα, IL-8 and IL-6 levels (Figure 9B-C). These clinical data validate the strong link between vascular ANGPTL2 and the marker of senescence p21, in yet non-atherosclerotic arteries, independently of age. These data also suggest that accumulation of senescent vascular ANGPTL2+;p21+ cells might precede plaque growth in human arteries.
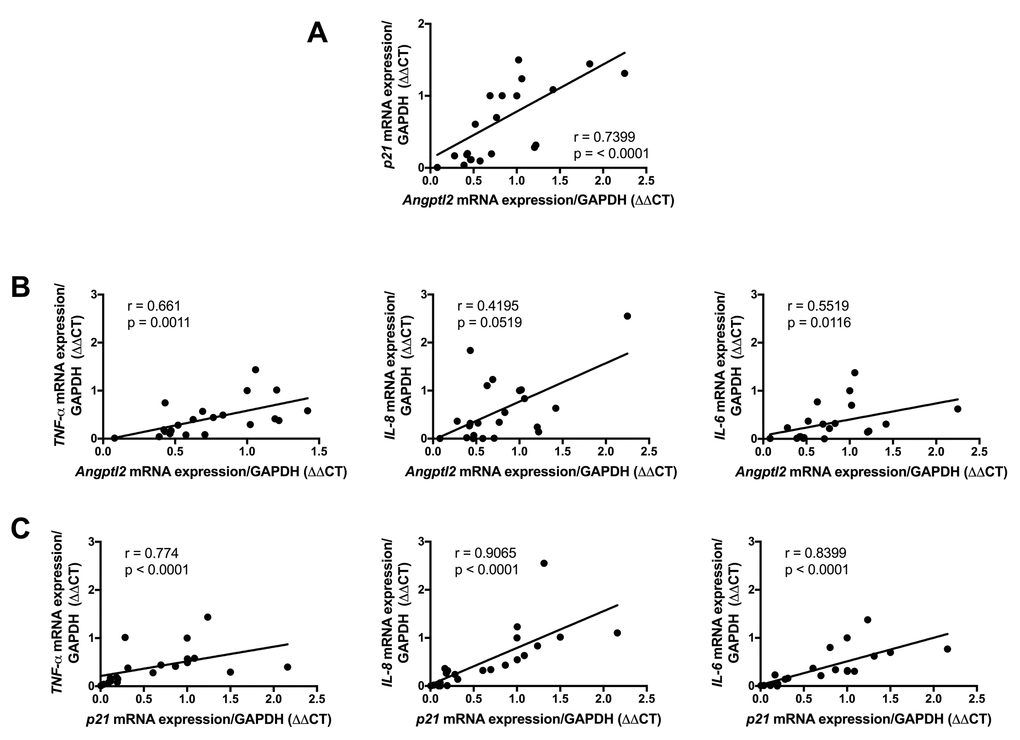
Figure 9. Association of vascular ANGPTL2, senescence and inflammation in IMA from atherosclerotic patients undergoing coronary artery by-pass surgery. Linear correlations between (A) ANGPTL2 and p21 mRNA expression, (B) ANGPTL2 and TNF-α,IL-8 and IL-6 mRNA expression and (C) p21 and TNF-α,IL-8 and IL-6 mRNA expression in IMA segments (n=26). Non-parametric Spearman correlation test was applied. Patient’s characteristics are described in Table S2.
Discussion
The novel findings of this study are that down-regulation of vascular Angptl2 may limit the progression of atherosclerotic lesions in pre-atherosclerotic dyslipidemic mice by 1) inducing clearance of senescent ECs through apoptosis and 2) by promoting endothelium repair and thus, attenuating the pro-inflammatory environment in the wall of the aorta. We have also shown that vascular ANGPTL2 expression is correlated with senescence and inflammatory cytokines of the SASP in pre-atherosclerotic internal mammary artery from patients with coronary artery disease, suggesting that angptl2 is a clinical marker of arterial senescence and that arterial senescence may precede plaque growth in human arteries. Altogether, these observations demonstrate that targeting vascular angptl2 is an innovative strategy to delay atherosclerosis by reducing endothelial senescence (Figure 10).
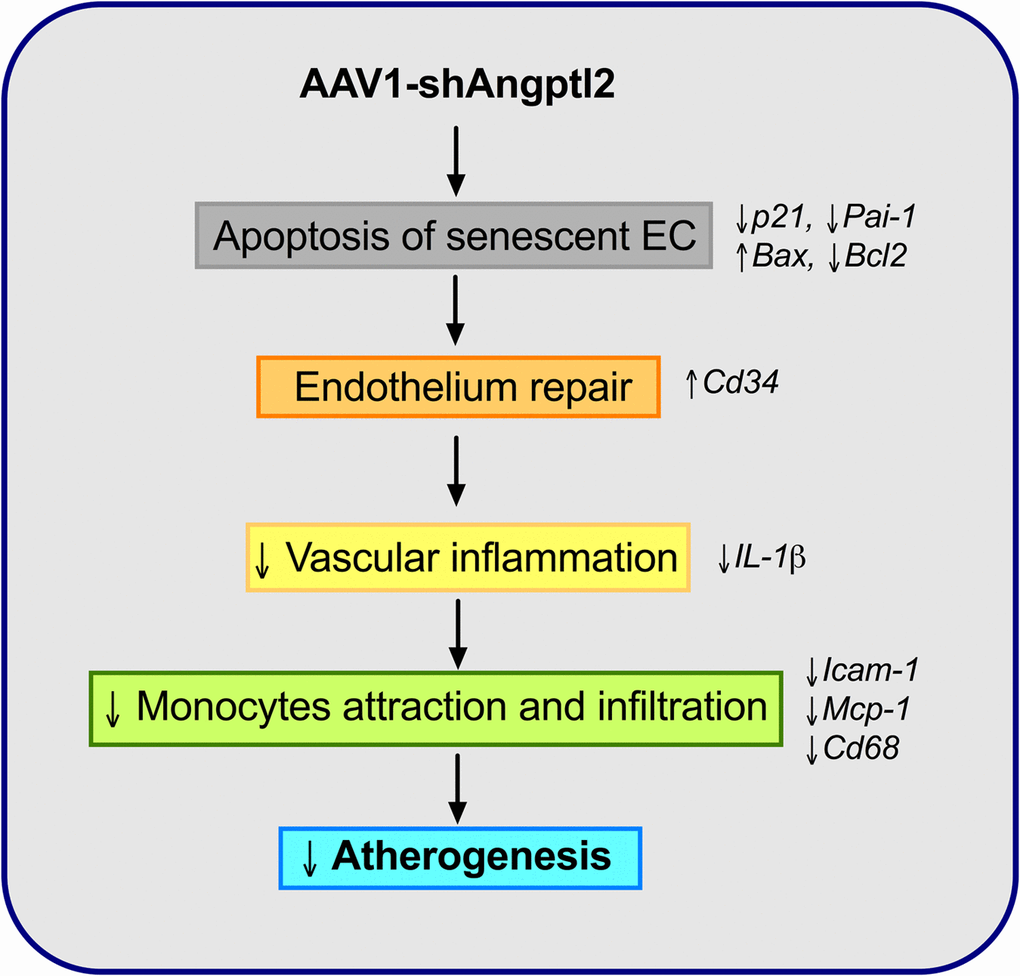
Figure 10. Schematic representation of the putative anti-atherogenic action of AAV1-shAngptl2. Elimination of angptl2+:p21+ senescent endothelial cells by apoptosis promotes endothelial repair. This leads to a reduced inflammatory profile, a lower immune cell adhesiveness and infiltration and therefore, a reduced atherogenicity.
Age-related atherosclerosis is partly driven by the accumulation of senescent cells in mice [12,14], creating a pro-inflammatory environment through their SASP [1]. Targeting senescent cells using senolytic drugs appears therefore a promising strategy to reduce the burden of age-related chronic inflammatory diseases [15], including atherosclerosis [16,17]. Selective and safe senolytics have yet to be discovered as most come from the oncology therapeutic field [18]. The protein angptl2, on the other hand, is highly expressed by senescent vascular human EC [30], but not by quiescent or proliferative EC [31], and is highly atherogenic when infused in young LDLr-/-;hApoB100-/- atherosclerotic mice [31]. Abundant ANGPTL2 expression was also observed in atheroma plaques of patients, particularly in EC and infiltrated macrophages [32]. The presence of senescent cells was also reported in human atherosclerotic arteries [17,41]. Although co-expression of vascular angptl2 and senescence markers has never been reported, our data show that by targeting angptl2+ vascular cells, senescent cells are eliminated, and thus strongly suggest that angptl2 is a marker of arterial senescence. Indeed, our recent study demonstrated that the per-operative variations in circulating plasma levels of angptl2 following cardiac surgery were determined by fat and arterial tissue inflammatory and senescence load of the patients [36]. Therefore, angptl2 is a clear marker of aging, which levels of expression are magnified by the presence of risk factors for vascular diseases [42] and are linked to arterial senescence [36]. The demonstration that down-regulation of angptl2 is anti-atherogenic therefore validates the concept of the contribution of senescent EC in the pathogenesis of atherosclerosis.
Several data collected in this work converge to support the contribution of angptl2+ senescent EC in endothelial dysfunction and in atherogenesis. Pro-inflammatory senescent EC are activated and promote aggregation of leukocytes [37], the initiating step of atherogenesis [38]. Our results reveal that following shAngptl2 treatment, the endothelial expression of the adhesion molecule Icam-1 and the endothelial expression of monocyte chemo-attractant protein Mcp1 were reduced, likely contributing to lower adhesion of Cd68+ monocytes onto the endothelium. Monocytes adhesion is crucial for macrophage infiltration and atheroma growth [38]. Endothelial cells and macrophages are the primary cells that secrete angptl2 in the atherosclerotic lesion [32], and angptl2 activates macrophage recruitment [29]. Accordingly, reducing angptl2+ senescent EC burden translates into lower EC activation, i.e. preserved endothelial anti-adhesive functions. Similarly, in hypercholesterolemic mice treated with the senolytic cocktail dasatinib/quercitin, endothelial dilatory function was improved via a greater nitric oxide (NO) bioavailability [12]. Interestingly, we reported that the dilatory eNOS-NO pathway was up-regulated in angptl2 knockdown mice [43], and that in contrast, angptl2 induced endothelial dysfunction [44]. Senescence was not quantified in the latter studies [43,44], but altogether our data suggest that lowering angptl2, and thus senescence, protects the endothelial functions. In the present study, p21 expression was not altered in the media of the aorta 3 months post-infection, suggesting that in this mouse model and at this young age, arterial smooth muscle cells, fibroblasts and foam cells are not senescent. In contrast, Angptl2 and Cd68 expression in the media decreased 3 months post-infection, as did Pai-1, Il-1β and Mcp1. On one hand, this result suggests that Cd68+ macrophages express angptl2 and contribute to the local inflammation, as previously reported [29,32,40] and, on the other hand, that elimination of senescent angptl2+ EC reduced monocyte adhesion and macrophage infiltration. Macrophages appear unique as they may express p16 as part of a physiological response to immune stimuli rather than through senescence [19,45,46]. Hence, reduced adhesion and infiltration of macrophages suggest improved endothelial functions 3 months post-AAV1-shAngptl2 therapy. Beyond the known benefits of eliminating senescent cells [18], the concomitant reduction both in the endothelium and the media of Pai-1, a well recognized SASP member and inducer of senescence [47], is clinically relevant: genetically driven high circulating levels of PAI-1 correlate with the severity of atherosclerosis [48], while conversely, a null mutation in its gene reduces markers of biological aging and increases longevity in humans [49].
A high level of senescent cells limits tissue remodelling and repair [50]. In contrast, parallel to the reduced endothelial expression of angptl2 and p21, endothelial (but not in the media) pro-apoptotic Bax expression was significantly increased after one week of treatment, while that of the anti-apoptotic Bcl2 was significantly decreased. These data suggest that angptl2+;p21+ senescent EC were eliminated following activation of this apoptotic pathway, which has been shown to be an effective mean of remodelling [51]. Selective induction of death by activation of apoptosis of senescent cells is the common mechanism of action of senolytics [12,14,19,52]. Thus, elimination of senescent angptl2+ EC cells by apoptosis is a senolytic approach.
Since the shAngptl2 therapy is anti-atherogenic, preserves endothelial anti-adhesive functions and reduces arterial wall inflammation, endothelial repair should occur. One major mechanism of repair is the recruitment of CD34+ progenitor cells from the circulation, and more specifically endothelial progenitor cells for re-endothelialization in mice [53]. Conversely, a reduced number of circulating EpC has been associated with an increase in major advanced cardiac events in patients with CVD [54]. Two weeks after shAngptl2 treatment, Cd34 expression tended to increase in the endothelium (data not shown), it increased significantly after 4 weeks and was sustained up to 3 months, suggesting that the shAngptl2 therapy stimulated repair of the endothelium. Thus, apoptosis of angptl2+;p21+ senescent EC likely stimulated endothelial repair, at least partly by incorporating circulating EpC. To our knowledge, this result is the first study to imply that integration of EpC into the native endothelium contributes to a functional endothelial repair post-senolytic therapy. Down-regulating vascular angptl2 should be safe; angptl2-KO [29] and our Angptl2-KD mice are healthy and resist better to an inflammatory stress [44].
Senescent EC overlay the atheroma plaque in patients [17]. To validate the clinical relevance of ANGPTL2 as a marker of vascular senescence, and to assess whether vascular senescence precedes plaque growth in patients, we measured the expression of ANGPTL2 and markers of senescence in functional, not atherosclerotic internal mammary artery segments from patients with CVD. In these arterial segments, the expression of p21 and that of ANGPTL2 were tightly associated, independently of the age of the donor, demonstrating that accumulation of senescent vascular cells is driven by stress (risk factors for CVD) more than age per se, as we previously reported in cultured EC isolated from mammary arteries [55]. The strong levels of association of ANGPTL2 and p21 levels with the pro-inflammatory cytokines of the SASP, i.e., TNFα, IL-8 and IL-6 levels strengthen the concept that senescence-driven chronic low-grade inflammation ultimately triggers the appearance of the atheroma with time.
Limitations of the study: first, almost all data were provided on mRNA level. Unfortunately, there was not enough material in the native endothelium layer scraped from the mouse aorta to look at the protein level. Second, n number is relatively small and variable due to the limited and variable availability of endothelial mRNA extractions. Third, expression of p21 mRNA was unaffected by the shAngptl2 in the media, suggesting either that cells in the media were not senescent, as we propose in these young ATX mice, or, alternatively, that angptl2 is not central to the maintenance of senescence in cells other than endothelial cells. Finally, the underlying mechanism of elevated CD34+ progenitor cell recruitment after shAngptl2 treatment was not elucidated. Because on one hand, angptl2 is anti-apoptotic [56], and because on the other hand, the shAngptl2 induced apoptosis of endothelial cells, it is possible that the generation of apoptotic bodies act as chemoattractant for EpC [57,58]. This attractive hypothesis would need to be validated in specific experiments.
In conclusion, therapeutic down-regulation of vascular angptl2 leads to the clearance of senescent EC by apoptosis, stimulates endothelial repair, preserves endothelial functions and reduces atherosclerosis. ANGPTL2 and p21 expression are tightly associated in non-atherosclerotic human arteries, which further suggests that accumulation of arterial senescence precedes age-related atherosclerosis. Altogether, these data suggest that targeting vascular angptl2 could be a selective and safe senolytic strategy to delay or reduce atherogenesis.
Materials and Methods
Animal study
All animal experiments were performed in accordance with the “Guide for the Care and Use of Experimental Animals of the Canadian Council on Animal Care” and the “Guide for the Care and Use of Laboratory Animals” of the US National Institutes of Health (NIH Publication No. 85-23, revised 1996) and was approved by the Montreal Heart Institute Ethics Committee. Knockout/transgenic severely dyslipidemic LDLr-/-;hApoB+/+ (ATX) male mice were fed a normal diet. Mice were anaesthetized with 44 mg/kg ketamine and 2.2 mg/kg xylazine and ventilated. Thoracic aortas of ATX and WT mice were used to quantify atherosclerotic lesions and to assess mRNA expression of senescent markers, inflammatory markers, progenitor marker and apoptosis markers both in the freshly isolated native endothelium and in the aorta wall [31]. Hearts, livers and blood of ATX mice were used to validate the vascular tropism of the AAV1.
AAV1 production
The protocol was adapted from a previous study [39]. HEKT293T competent cells were plated until a confluence of 70%, and were then transfected overnight with 12 µg/mL of the pXYZ C1 plasmid vector (serotype 1), 4 µg/mL of the plasmid containing shAngptl2, shSCR (see shRNA sequences Table S1) or mCherry red fluorescent protein sequence, and 48 µg/mL of polyethylenimin (Sigma) in a starvation medium. The next day, the medium was changed and cells were incubated for 48h with normal growth medium. Then, cells were collected and lysed with Tris 1M/ NaCl 5M pH 8.5. Lysis was accelerated by several freeze/thaw steps and 1 µL of MgCl2 (1M) in addition to benzonase (250U/µL, Sigma) were added per mL of lysate. After centrifugation, AAV1 were isolated from the supernatant by iodixanol gradients during ultracentrifugation, then extracted with a syringe and concentrated in a PBST buffer volume between 0.25 ml and 0.5 ml with Ultracel-100 regenerated cellulose membrane (100 kDa, EMD Millipore).
AAV1 titration and administration in ATX mice
Titration was performed by qPCR reactions using a StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). AAV1 were quantified using a standard range made by serial dilutions (10 ng to 10-6 ng) of shAngptl2 and shSCR plasmids containing a target sequence (BGH) or the plasmid containing mCherry sequence. The primers for BGH target sequence and mCherry were designed using Clone Manager software (Table S3). Each mouse received a systemic (i.v.) bolus injection of 1011 AAV1 particles at 3-month old (-mo) and were studied at 6-mo; some mice received an injection at 5-mo and were studied after 1 week, 2 weeks or 4 weeks. A pilot study showed that injection of 5x1010 AAV1 particles was sub-optimal, leading to an inconsistent reduction in angptl2 by ∼50% (data not shown).
Quantification of atherosclerotic lesions
Freshly isolated thoracic aortas of ATX mice were longitudinally opened and pictures taken with a digital camera. Atherosclerosis lesions were quantified by measuring the white spots in the aorta using ImageJ software as previously [31]. Plaque areas were expressed as percentage of total aortic area.
Total RNA extraction
Total RNA was extracted from freshly isolated aorta, separately in both the endothelial layer and the arterial wall. Native ECs were gently scraped with the tip of a scalpel blade from longitudinally opened segments of the thoracic aorta and endothelial mRNA was extracted as previously [31]; the denuded aorta wall (including the atheroma plaque), the liver and the heart were pulverized in liquid nitrogen with a Cell crusher kit (Cellcrusher Limited). RNAs were extracted using an RNeasy Mini Kit (Qiagen). Contamination with genomic DNA was prevented by a digestion with a DNase I mix (Qiagen), according to the manufacturer’s guidelines. Total RNA was quantified using a NanoDrop ND-100 spectrophotometer.
Real-Time quantitative PCR
Total RNA was reverse transcribed into first-strand complementary DNA with M-MLV reverse transcriptase (Thermo Fisher), using random hexamer primers. The qPCR reactions were carried out on diluted RT products by using the DNA-binding dye SYBR Green PCR Master Mix (Thermo Fisher Scientific) to detect PCR products with StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). The primers of target genes (Angptl2, p21, Il-1β, Icam-1, Cd68, Cd34, Mcp1, Bax, Bcl2) were designed using Clone Manager software (Table S3 (mouse) and S4 (human)). All samples were run in duplicate and the fold changes in gene expression were calculated by a ΔΔCT method using cycloA (cyclophilin A) as the housekeeping gene. N numbers are not homogeneous due to the limited and variable availability of endothelial mRNA extraction.
Immunofluorescence
Fixed tissues (transversely aortic frozen sections) were incubated with 1:100 diluted rabbit anti-mCherry (#ab167453; Abcam) or with 1:200 diluted rabbit anti-CD34 (#ab185732; Abcam) and 1:800 diluted secondary antibody Alexa fluor-647 anti-rabbit (#A31573, Thermo Fisher Scientific). DNA counterstaining was performed by incubating fixed tissues with DAPI (D1306, Thermo Fisher Scientific). Fluorescence was visualized by confocal microscopy (Zeiss LSM 510) with 40X magnification for mCherry staining and 20X magnification for CD34 staining. CD34 staining was normalized to the total number of nuclei present in the endothelial layer (DAPI staining).
Biochemical analysis
Blood was rapidly collected after sacrifices and then centrifuged within minutes to collect the plasma. Samples were stored frozen in aliquots at -80°C until further analysis. Plasma metabolic profile (total cholesterol, glucose and triglycerides) was measured at the biochemical laboratory of the Montreal Heart Institute.
Human study
Twenty-six coronary artery disease (CAD) patients undergoing coronary arterial bypass (CABG, n=16), or both CABG and aortic or mitral valve replacement (VR) (n=10) between February 2014 and January 2016, were prospectively included in the study at the Montreal Heart Institute (Quebec, Canada). Inclusion criteria were patients undergoing elective CABG and VR with normal left ventricular function at pre-operative evaluation. Patients who underwent other type of procedure were excluded. All patients signed an informed consent and the study (#ICM 13-1492) was approved by the ethical committee on human research of the Montreal Heart Institute. On the overall population, the mean age was 67±8 years and 92% were male. All patients had at least one risk factor for CVD and received preventive treatment such aspirin or statin before surgery. Left ventricular function was normal in all patients. The basal pre-operative characteristics of the patients are detailed in the Table S2. All patients underwent cardiac surgery with cardiopulmonary bypass (CPB). During surgery, a distal segment of left internal mammary artery (IMA) measuring at least 1 cm in length was harvested and stored at -80°C for all patients. Then gene expression of ANGPTL2 and other markers of inflammation (TNFα, IL-8, IL-6) and senescence (p21) were measured in this tissue. Frozen IMA fragments were pulverized in liquid nitrogen and dry ice with a Cell crusher kit (Cellcrusher Limited). Samples of organ powder were then processed for RNA extraction according to the manufacturer's instructions (RNeasy mini kit. Qiagen). Contamination with DNA was prevented by a digestion with a DNase I mix (Qiagen) according to the manufacturer’s guidelines. Total RNA was quantified using a NanoDrop ND-100 spectrophotometer. Total RNA was reverse-transcribed into first-strand complementary DNA with M-MLV reverse transcriptase (Thermo Fisher Scientific) using random hexamer primers. The primers of target genes were designed using Clone Manager software (Table S4). Quantitative polymerase chain reaction was performed using the SYBR Green PCR Master Mix (Thermo Fisher Scientific). All samples were run in duplicate and the fold changes in gene expression were calculated by a ΔΔCT method using GAPDH as the housekeeping gene.
Statistical analysis
Data are presented as mean±SEM, with “n” indicating the number of animals or patients. Animal data were analyzed using parametric and non-parametric tests: a two-tailed unpaired t test was used to compare plaque growth and endothelial gene (Angptl2,p21 and Pai-1) expression in ATX mice at 3- vs. 12-mo (Figure 1). A two-way ANOVA with Sidak’s multiple comparisons test was performed to compare mRNA expressions in WT and ATX mice (Figure 2). One-way ANOVA with Tukey’s posthoc-test was used to compare plaque area in untreated 3-mo, AAV1-treated or -untreated 6-mo ATX mice (Figure 4). Unpaired t test or Mann Whitney test was used to compare mRNA expression in shSCR group (control) versus shAngptl2 group (Figures 5, 6, 7 and 8). Human data are presented as dot plots. Non-parametric Spearman correlation analyses were performed to correlate the mRNA levels of ANGPTL2 and p21 with TNFα, IL-8 and IL-6 in IMA segments from patients (Figure 9). All statistics were performed using Graph Pad Prism 7.0.
Supplementary Materials
Abbreviations
AAV1: adeno-associated virus serotype 1; ACEi: angiotensin converting enzyme inhibitors; Angptl2: angiopoietin-like 2: ARA, angiotensin II receptor antagonists; ATX: dyslipidemic LDLr-/-; ApoB100+/+: spontaneously atherosclerotic mice; Bax: Bcl-2-associated X protein; Bcl2: B-cell lymphoma 2; BMI: body mass index; CAD: coronary artery disease; CD34: Cluster of Differentiation 34; CD68: Cluster of Differentiation 68; COPD: chronic obstructive pulmonary disease; CVD: cardiovascular diseases; EC: endothelial cells; HbA1C: glycated hemoglobin A1C; ICAM-1: intracellular adhesion molecule-1; IL-1β: interleukin-1β; IL-6: interleukin-6; IL-8: interleukin-8; MCP-1: monocyte chemoattractant protein 1; p21: cyclin-dependent kinase inhibitor 1; PAI-1: plasminogen activator inhibitor 1; SASP: senescence-associated secretory phenotype; shRNA: small hairpin ribonucleic acid; SnC: senescent cells; SCR: scramble sequence; TNF-α: tumor necrosis factor alpha.
Author Contributions
LC and ET were responsible for the experimental design. LC carried out experiments and data analysis in mice. PL carried out quantitative PCR analysis in the human samples. PL and NTT analyzed the data of the human samples. MM designed the plasmids and all the primers. LV carried out confocal microscopy. PEN and MC performed the cardiac surgeries and supervised analysis of the human samples. LC, PL, NTT and ET wrote the manuscript. GF edited the manuscript. All authors reviewed the manuscript.
Acknowledgements
We thank Natacha Duquette and Marie-Ève Higgins for expert assistance in injecting the AAV1-shRNA in mice.
Conflicts of Interest
The authors declare that they have no conflicts of interest in relation to this work.
Funding
This research was funded by the Canadian Institute for Health Research (MOP133649, E.T.) and the Foundation of the Montreal Heart Institute (E.T.).
References
- 1. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010; 5:99–118. https://doi.org/10.1146/annurev-pathol-121808-102144 [PubMed]
- 2. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D, Hohmeyer A, Gereke M, Rudalska R, Potapova A, Iken M, Vucur M, Weiss S, et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011; 479:547–51. https://doi.org/10.1038/nature10599 [PubMed]
- 3. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007; 445:656–60. https://doi.org/10.1038/nature05529 [PubMed]
- 4. Deschênes-Simard X, Gaumont-Leclerc MF, Bourdeau V, Lessard F, Moiseeva O, Forest V, Igelmann S, Mallette FA, Saba-El-Leil MK, Meloche S, Saad F, Mes-Masson AM, Ferbeyre G. Tumor suppressor activity of the ERK/MAPK pathway by promoting selective protein degradation. Genes Dev. 2013; 27:900–15. https://doi.org/10.1101/gad.203984.112 [PubMed]
- 5. Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS. BRAF(E600) in benign and malignant human tumours. Oncogene. 2008; 27:877–95. https://doi.org/10.1038/sj.onc.1210704 [PubMed]
- 6. Castro P, Xia C, Gomez L, Lamb DJ, Ittmann M. Interleukin-8 expression is increased in senescent prostatic epithelial cells and promotes the development of benign prostatic hyperplasia. Prostate. 2004; 60:153–59. https://doi.org/10.1002/pros.20051 [PubMed]
- 7. Biran A, Zada L, Abou Karam P, Vadai E, Roitman L, Ovadya Y, Porat Z, Krizhanovsky V. Quantitative identification of senescent cells in aging and disease. Aging Cell. 2017; 16:661–71. https://doi.org/10.1111/acel.12592 [PubMed]
- 8. Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, Khazaie K, Miller JD, van Deursen JM. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature. 2016; 530:184–89. https://doi.org/10.1038/nature16932 [PubMed]
- 9. Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, van Deursen JM. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. 2011; 479:232–36. https://doi.org/10.1038/nature10600 [PubMed]
- 10. Baar MP, Brandt RM, Putavet DA, Klein JD, Derks KW, Bourgeois BR, Stryeck S, Rijksen Y, van Willigenburg H, Feijtel DA, van der Pluijm I, Essers J, van Cappellen WA, et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell. 2017; 169:132–147.e16. https://doi.org/10.1016/j.cell.2017.02.031 [PubMed]
- 11. Yosef R, Pilpel N, Tokarsky-Amiel R, Biran A, Ovadya Y, Cohen S, Vadai E, Dassa L, Shahar E, Condiotti R, Ben-Porath I, Krizhanovsky V. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun. 2016; 7:11190. https://doi.org/10.1038/ncomms11190 [PubMed]
- 12. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, Hagler M, Jurk D, Smith LA, Casaclang-Verzosa G, Zhu Y, Schafer MJ, Tchkonia T, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. 2016; 15:973–77. https://doi.org/10.1111/acel.12458 [PubMed]
- 13. Minamino T, Yoshida T, Tateno K, Miyauchi H, Zou Y, Toko H, Komuro I. Ras induces vascular smooth muscle cell senescence and inflammation in human atherosclerosis. Circulation. 2003; 108:2264–69. https://doi.org/10.1161/01.CIR.0000093274.82929.22 [PubMed]
- 14. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. 2016; 354:472–77. https://doi.org/10.1126/science.aaf6659 [PubMed]
- 15. McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol. 2018; 217:65–77. https://doi.org/10.1083/jcb.201708092 [PubMed]
- 16. Katsuumi G, Shimizu I, Yoshida Y, Minamino T. Vascular Senescence in Cardiovascular and Metabolic Diseases. Front Cardiovasc Med. 2018; 5:18. https://doi.org/10.3389/fcvm.2018.00018 [PubMed]
- 17. Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002; 105:1541–44. https://doi.org/10.1161/01.CIR.0000013836.85741.17 [PubMed]
- 18. Childs BG, Gluscevic M, Baker DJ, Laberge RM, Marquess D, Dananberg J, van Deursen JM. Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov. 2017; 16:718–35. https://doi.org/10.1038/nrd.2017.116 [PubMed]
- 19. Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM, Inman CL, Ogrodnik MB, Hachfeld CM, Fraser DG, Onken JL, Johnson KO, Verzosa GC, et al. Senolytics improve physical function and increase lifespan in old age. Nat Med. 2018; 24:1246–56. https://doi.org/10.1038/s41591-018-0092-9 [PubMed]
- 20. Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M, O’Hara SP, LaRusso NF, Miller JD, et al. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell. 2015; 14:644–58. https://doi.org/10.1111/acel.12344 [PubMed]
- 21. Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, Luo Y, Wang X, Aykin-Burns N, et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med. 2016; 22:78–83. https://doi.org/10.1038/nm.4010 [PubMed]
- 22. Pan J, Li D, Xu Y, Zhang J, Wang Y, Chen M, Lin S, Huang L, Chung EJ, Citrin DE, Wang Y, Hauer-Jensen M, Zhou D, Meng A. Inhibition of Bcl-2/xl With ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int J Radiat Oncol Biol Phys. 2017; 99:353–61. https://doi.org/10.1016/j.ijrobp.2017.02.216 [PubMed]
- 23. Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature. 2018; 562:578–82. https://doi.org/10.1038/s41586-018-0543-y [PubMed]
- 24. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP, Athineos D, Kang TW, Lasitschka F, Andrulis M, Pascual G, Morris KJ, Khan S, et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013; 15:978–90. https://doi.org/10.1038/ncb2784 [PubMed]
- 25. Shimamoto A, Kagawa H, Zensho K, Sera Y, Kazuki Y, Osaki M, Oshimura M, Ishigaki Y, Hamasaki K, Kodama Y, Yuasa S, Fukuda K, Hirashima K, et al. Reprogramming suppresses premature senescence phenotypes of Werner syndrome cells and maintains chromosomal stability over long-term culture. PLoS One. 2014; 9:e112900. https://doi.org/10.1371/journal.pone.0112900 [PubMed]
- 26. Nagano T, Nakano M, Nakashima A, Onishi K, Yamao S, Enari M, Kikkawa U, Kamada S. Identification of cellular senescence-specific genes by comparative transcriptomics. Sci Rep. 2016; 6:31758. https://doi.org/10.1038/srep31758 [PubMed]
- 27. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S, Medina-Echeverz J, Longerich T, Forgues M, Reisinger F, Heikenwalder M, Wang XW, Zender L, Greten TF. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016; 30:533–47. https://doi.org/10.1016/j.ccell.2016.09.003 [PubMed]
- 28. Pienimaeki-Roemer A, Konovalova T, Musri MM, Sigruener A, Boettcher A, Meister G, Schmitz G. Transcriptomic profiling of platelet senescence and platelet extracellular vesicles. Transfusion. 2017; 57:144–56. https://doi.org/10.1111/trf.13896 [PubMed]
- 29. Tabata M, Kadomatsu T, Fukuhara S, Miyata K, Ito Y, Endo M, Urano T, Zhu HJ, Tsukano H, Tazume H, Kaikita K, Miyashita K, Iwawaki T, et al. Angiopoietin-like protein 2 promotes chronic adipose tissue inflammation and obesity-related systemic insulin resistance. Cell Metab. 2009; 10:178–88. https://doi.org/10.1016/j.cmet.2009.08.003 [PubMed]
- 30. Farhat N, Thorin-Trescases N, Voghel G, Villeneuve L, Mamarbachi M, Perrault LP, Carrier M, Thorin E. Stress-induced senescence predominates in endothelial cells isolated from atherosclerotic chronic smokers. Can J Physiol Pharmacol. 2008; 86:761–69. https://doi.org/10.1139/Y08-082 [PubMed]
- 31. Farhat N, Thorin-Trescases N, Mamarbachi M, Villeneuve L, Yu C, Martel C, Duquette N, Gayda M, Nigam A, Juneau M, Allen BG, Thorin E. Angiopoietin-like 2 promotes atherogenesis in mice. J Am Heart Assoc. 2013; 2:e000201. https://doi.org/10.1161/JAHA.113.000201 [PubMed]
- 32. Horio E, Kadomatsu T, Miyata K, Arai Y, Hosokawa K, Doi Y, Ninomiya T, Horiguchi H, Endo M, Tabata M, Tazume H, Tian Z, Takahashi O, et al. Role of endothelial cell-derived angptl2 in vascular inflammation leading to endothelial dysfunction and atherosclerosis progression. Arterioscler Thromb Vasc Biol. 2014; 34:790–800. https://doi.org/10.1161/ATVBAHA.113.303116 [PubMed]
- 33. Thorin-Trescases N, Hayami D, Yu C, Luo X, Nguyen A, Larouche JF, Lalongé J, Henri C, Arsenault A, Gayda M, Juneau M, Lambert J, Thorin E, Nigam A. Exercise Lowers Plasma Angiopoietin-Like 2 in Men with Post-Acute Coronary Syndrome. PLoS One. 2016; 11:e0164598. https://doi.org/10.1371/journal.pone.0164598 [PubMed]
- 34. Gellen B, Thorin-Trescases N, Sosner P, Gand E, Saulnier PJ, Ragot S, Fraty M, Laugier S, Ducrocq G, Montaigne D, Llaty P, Rigalleau V, Zaoui P, et al. ANGPTL2 is associated with an increased risk of cardiovascular events and death in diabetic patients. Diabetologia. 2016; 59:2321–30. https://doi.org/10.1007/s00125-016-4066-5 [PubMed]
- 35. Yosef R, Pilpel N, Papismadov N, Gal H, Ovadya Y, Vadai E, Miller S, Porat Z, Ben-Dor S, Krizhanovsky V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017; 36:2280–95. https://doi.org/10.15252/embj.201695553 [PubMed]
- 36. Noly PE, Labbé P, Thorin-Trescases N, Fortier A, Nguyen A, Thorin E, Carrier M. Reduction of plasma angiopoietin-like 2 after cardiac surgery is related to tissue inflammation and senescence status of patients. J Thorac Cardiovasc Surg. 2019. Epub ahead of print. https://doi.org/10.1016/j.jtcvs.2018.12.047 [PubMed]
- 37. Erusalimsky JD. Vascular endothelial senescence: from mechanisms to pathophysiology. J Appl Physiol (1985). 2009; 106:326–32. https://doi.org/10.1152/japplphysiol.91353.2008 [PubMed]
- 38. Gimbrone MA
Jr , García-Cardeña G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ Res. 2016; 118:620–36. https://doi.org/10.1161/CIRCRESAHA.115.306301 [PubMed] - 39. Chen S, Kapturczak M, Loiler SA, Zolotukhin S, Glushakova OY, Madsen KM, Samulski RJ, Hauswirth WW, Campbell-Thompson M, Berns KI, Flotte TR, Atkinson MA, Tisher CC, Agarwal A. Efficient transduction of vascular endothelial cells with recombinant adeno-associated virus serotype 1 and 5 vectors. Hum Gene Ther. 2005; 16:235–47. https://doi.org/10.1089/hum.2005.16.235 [PubMed]
- 40. Tazume H, Miyata K, Tian Z, Endo M, Horiguchi H, Takahashi O, Horio E, Tsukano H, Kadomatsu T, Nakashima Y, Kunitomo R, Kaneko Y, Moriyama S, et al. Macrophage-derived angiopoietin-like protein 2 accelerates development of abdominal aortic aneurysm. Arterioscler Thromb Vasc Biol. 2012; 32:1400–09. https://doi.org/10.1161/ATVBAHA.112.247866 [PubMed]
- 41. Minamino T, Miyauchi H, Yoshida T, Komuro I. Endothelial cell senescence in human atherosclerosis: role of telomeres in endothelial dysfunction. J Cardiol. 2003; 41:39–40. [PubMed]
- 42. Thorin-Trescases N, Thorin E. High Circulating Levels of ANGPTL2: Beyond a Clinical Marker of Systemic Inflammation. Oxid Med Cell Longev. 2017; 2017:1096385. https://doi.org/10.1155/2017/1096385 [PubMed]
- 43. Yu C, Luo X, Duquette N, Thorin-Trescases N, Thorin E. Knockdown of angiopoietin like-2 protects against angiotensin II-induced cerebral endothelial dysfunction in mice. Am J Physiol Heart Circ Physiol. 2015; 308:H386–97. https://doi.org/10.1152/ajpheart.00278.2014 [PubMed]
- 44. Yu C, Luo X, Farhat N, Daneault C, Duquette N, Martel C, Lambert J, Thorin-Trescases N, Rosiers CD, Thorin E. Lack of angiopoietin-like-2 expression limits the metabolic stress induced by a high-fat diet and maintains endothelial function in mice. J Am Heart Assoc. 2014; 3:e001024. https://doi.org/10.1161/JAHA.114.001024 [PubMed]
- 45. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin II, Leonova KI, Consiglio CR, Gollnick SO, et al. p16(Ink4a) and senescence-associated β-galactosidase can be induced in macrophages as part of a reversible response to physiological stimuli. Aging (Albany NY). 2017; 9:1867–84. https://doi.org/10.18632/aging.101268 [PubMed]
- 46. Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP, Rydkina E, Vujcic S, Balan K, Gitlin I, Leonova K, Polinsky A, Chernova OB, Gudkov AV. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY). 2016; 8:1294–315. https://doi.org/10.18632/aging.100991 [PubMed]
- 47. Vaughan DE, Rai R, Khan SS, Eren M, Ghosh AK. Plasminogen Activator Inhibitor-1 Is a Marker and a Mediator of Senescence. Arterioscler Thromb Vasc Biol. 2017; 37:1446–52. https://doi.org/10.1161/ATVBAHA.117.309451 [PubMed]
- 48. Cvetkovic D, Lafaro R, Giamelli J, Suvro S, Erb M, Yaghoubian S. Quadruple Vessel Coronary Artery Bypass Grafting in a 14-Year-Old Child With Plasminogen Activator Inhibitor-1 4G/4G Gene Polymorphism. Semin Cardiothorac Vasc Anesth. 2016; 20:163–67. https://doi.org/10.1177/1089253216631426 [PubMed]
- 49. Khan SS, Shah SJ, Klyachko E, Baldridge AS, Eren M, Place AT, Aviv A, Puterman E, Lloyd-Jones DM, Heiman M, Miyata T, Gupta S, Shapiro AD, Vaughan DE. A null mutation in SERPINE1 protects against biological aging in humans. Sci Adv. 2017; 3:o1617. https://doi.org/10.1126/sciadv.aao1617 [PubMed]
- 50. van Deursen JM. The role of senescent cells in ageing. Nature. 2014; 509:439–46. https://doi.org/10.1038/nature13193 [PubMed]
- 51. Westphal D, Dewson G, Czabotar PE, Kluck RM. Molecular biology of Bax and Bak activation and action. Biochim Biophys Acta. 2011; 1813:521–31. https://doi.org/10.1016/j.bbamcr.2010.12.019 [PubMed]
- 52. Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, Stout MB, Pirtskhalava T, Giorgadze N, Johnson KO, Giles CB, Wren JD, Niedernhofer LJ, Robbins PD, Kirkland JL. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell. 2016; 15:428–35. https://doi.org/10.1111/acel.12445 [PubMed]
- 53. Nowak G, Karrar A, Holmén C, Nava S, Uzunel M, Hultenby K, Sumitran-Holgersson S. Expression of vascular endothelial growth factor receptor-2 or Tie-2 on peripheral blood cells defines functionally competent cell populations capable of reendothelialization. Circulation. 2004; 110:3699–707. https://doi.org/10.1161/01.CIR.0000143626.16576.51 [PubMed]
- 54. Werner N, Kosiol S, Schiegl T, Ahlers P, Walenta K, Link A, Böhm M, Nickenig G. Circulating endothelial progenitor cells and cardiovascular outcomes. N Engl J Med. 2005; 353:999–1007. https://doi.org/10.1056/NEJMoa043814 [PubMed]
- 55. Voghel G, Thorin-Trescases N, Farhat N, Nguyen A, Villeneuve L, Mamarbachi AM, Fortier A, Perrault LP, Carrier M, Thorin E. Cellular senescence in endothelial cells from atherosclerotic patients is accelerated by oxidative stress associated with cardiovascular risk factors. Mech Ageing Dev. 2007; 128:662–71. https://doi.org/10.1016/j.mad.2007.09.006 [PubMed]
- 56. Horiguchi H, Endo M, Miyamoto Y, Sakamoto Y, Odagiri H, Masuda T, Kadomatsu T, Tanoue H, Motokawa I, Terada K, Morioka MS, Manabe I, Baba H, Oike Y. Angiopoietin-like protein 2 renders colorectal cancer cells resistant to chemotherapy by activating spleen tyrosine kinase-phosphoinositide 3-kinase-dependent anti-apoptotic signaling. Cancer Sci. 2014; 105:1550–59. https://doi.org/10.1111/cas.12554 [PubMed]
- 57. Hristov M, Erl W, Linder S, Weber PC. Apoptotic bodies from endothelial cells enhance the number and initiate the differentiation of human endothelial progenitor cells in vitro. Blood. 2004; 104:2761–66. https://doi.org/10.1182/blood-2003-10-3614 [PubMed]
- 58. Sabatier F, Camoin-Jau L, Anfosso F, Sampol J, Dignat-George F. Circulating endothelial cells, microparticles and progenitors: key players towards the definition of vascular competence. J Cell Mol Med. 2009; 13:454–71. https://doi.org/10.1111/j.1582-4934.2008.00639.x [PubMed]